
Abstract
We report the mitogenomes for five species of the Ischnocnema guentheri series, being the first described for this genus of brachycephalid frogs. We assembled mitogenomes from anchored hybrid enrichment data and recovered the 13 protein-coding genes, 22 tRNA genes, and two rRNA genes for all species. The general structure agrees with most previously sequenced neobatrachians, with two exceptions: the origin of replication of L-strand (OL) was found between tRNA-A and tRNA-N, and the position of tRNA-L and tRNA-T, which are dispersed in the control region. We provide a phylogenetic tree with outgroups, which is consistent with previous phylogenetic hypotheses.
The Neotropical genus Ischnocnema (Reinhardt and Lütken Citation1862) comprises 37 species (Frost Citation2018) of leaf-litter dwelling frogs divided into five species series and three species unassigned to any series (Taucce at al. Citation2018). Within this genus, the I. guentheri series comprises 10 species distributed all over the southern and central portions of the Atlantic Forest, throughout seven Brazilian states and the Argentinean province of Misiones (Frost Citation2018). The series has a challenging taxonomy, with notable intra and inter-specific morphological variation (Heyer Citation1984), and some of its members may actually represent complexes of morphologically cryptic species (Gehara et al. Citation2013). Herein, we provide complete or nearly complete metagenome sequences for half (five) of the currently recognized species of the I. guentheri series, assembled from anchored hybrid enrichment data (Lemmon et al. 2012): I. erythromera (Heyer Citation1984), I. guentheri (Steindachner Citation1864), I. nasuta (Lutz Citation1925), I. oea (Heyer Citation1984) (one specimen each), and I. henselii (Peters Citation1870) (five specimens). Voucher specimens and tissue samples are housed in the CFBH or LGE collections (acronyms follow Sabaj Citation2016).
We extracted total DNA from ethanol-preserved muscle or liver tissues using the DNeasy Qiagen® kit following manufacturer’s protocols. DNA was eluted to a volume of 100 μl and quantified using a Qubit fluorometer dsDNA BR Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA). Extractions were sent to the Center for Anchored Phylogenomics (Tallahassee, FL) to be sequenced with a method for anchored hybrid enrichment analysis (Lemmon et al. Citation2012). Samples were pooled after indexing and hybrid enrichments were performed with probes designed for anchored loci from amphibians (Barrow et al. Citation2018; Heinicke et al. Citation2018). Mitochondrial sequences were recovered as bycatch during the process of hybrid enrichment. Sequencing was carried out on an Illumina HiSeq2500 sequencer. For mitochondrial genomes assemblies, each lane of raw sequence reads was first concatenated per sample and quality-trimmed using Trimmomatic (Bolger et al. Citation2014). Then, we used MITObim v1.9 (Hahn et al. Citation2013) using as reference the mitogenome of Eleutherodactylus atkinsi (GenBank number: JX564864) to assemble the mitogenome of Ischnocnema oea. Next, we checked the quality and coverage of this new mitogenome and used it to assemble the remaining specimens of Ischnocnema. Assemblies were checked for quality by mapping the mitochondrial reads recovered by MITObim to the final fasta file with Geneious R11 (Kearse et al. Citation2012). We also used Geneious R11 to test for mitogenome circularity and completeness using the ‘De novo assemble’ tool. Regions with low coverage (less than eight reads) were manually edited to unknown nucleotides (‘N’). The preliminary annotation of final mitochondrial genomes was carried out by MITOS 2 (Bernt, Donath, Jühling, et al. Citation2013), available online at http://mitos2.bioinf.uni-leipzig.de/index.py, and verified by alignment with Eleutherodactylus atkinsi. The protein-coding regions were checked to confirm no indels or stop codons were present. The new mitogenomes have been deposited in GenBank under accession numbers MH492729–MH492737.
We recovered the typical 13 protein-coding genes, two rRNA genes and all 22 tRNA genes for all specimens, except I. guentheri. The mitogenome sequences of I. nasuta, one specimen of I. henselii and I. guentheri are complete and circular, but we could not annotate the tRNA-T in I. guentheri, probably because it is in a low coverage region. Other specimens are incomplete only in the control region (Supplementary Table S1; available at https://figshare.com/s/1537a00d5bf67288030e). Generally, the gene order observed agrees with most previously sequenced neobatrachian frogs (Zhang et al. Citation2013), with two main exceptions: the position of the putative origin of replication of the L-strand (OL), which we found between tRNA-A and tRNA-N (cluster WA(OL)NCY) and not between tRNA-N and tRNA-C (cluster WAN(OL)CY); and the organization of the LTPF cluster in the 5′ of the 12S rRNA gene, which differs in the Ischnocnema by the tRNA-L and tRNA-T being located within the control region (see Supplementary Figure S1). We also found a new arrangement for the cluster LTPF in I. erythromera, in which the tRNA arrangement is PLTF. These tRNAs clusters are known to be a hotspot of gene rearrangement in amphibians (San Mauro et al. Citation2006; Bernt, Braband, et al. Citation2013; Zhang et al. Citation2013) and the rearrangements in these regions are normally interpreted as a result of replication errors near the replication origins O-L and O-H (Kurabayashi and Sumida Citation2013).
Table 1. GenBank accession numbers, collection numbers, local of collection, and geospatial coordinates of specimens of the Ischnocnema, Gastrotheca, Pristimantis, and Eleutherodactylus species used in this study.
For phylogenetic inference, Ischnocnema sequences were aligned to publish complete or near complete genomes of four outgroups () using the software MAFFT v.7 (Katoh and Standley Citation2013). To avoid ambiguous alignments, we used only protein-coding and rRNA genes in the analyses. Search for the best partition scheme and best fitting nuclear substitution models was performed with PartitionFinder 2.1.1 (Lanfear et al. Citation2017). Phylogenetic analyses were performed under Bayesian inference (BI), maximum likelihood (ML), and maximum parsimony (MP) optimality criteria with the software MrBayes 3.2.6 (Ronquist et al. Citation2012), RAxML 8.2.11 (Stamatakis Citation2014), and TNT 1.5 (Goloboff and Catalano Citation2016), respectively. The best partition scheme with respective best-fitting substitution models and details on each phylogenetic analysis are given in Supplemental Online Material.
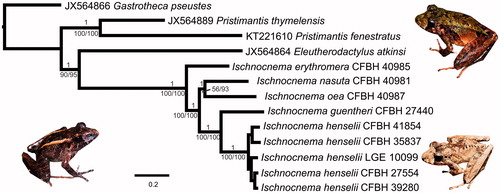
The three phylogenetic analyses are congruent and show all Ischnocnema species as a fully supported clade which is placed with high support as sister group to Eleutherodactylus atkinsi (). The two species of Pristimantis (Jiménez de la Espada Citation1870) also appear as a fully supported clade. Ischnocnema erythromera is the sister species of all other Ischnocnema in our tree and I. guentheri and I. henselii, as well as I. nasuta and I. oea, appear, respectively, as sister species. The only clade in our phylogeny that did not receive strong support was I. nasuta + I. oea, but only in the ML analysis (56% of bootstrap replicates). These results are congruent with the previous phylogenetic hypothesis encompassing all these Ischnocnema species based on a selection of mitochondrial and nuclear genes (Canedo and Haddad Citation2012 Taucce et al. Citation2018). The mitogenomes assembled here provide important information regarding the relationships within the I. guentheri species series and their genomic evolution.
Figure 1. The 50% majority rule consensus tree from Bayesian inference analysis of mitogenomic sequences of Ischnocnema and four outgroups. Numbers above branches are posterior probabilities and numbers below branches are maximum likelihood bootstrap replicates (left) and maximum parsimony jackknife replicates (right). No support below species level is shown. Pictures show Ischnocnema nasuta (left), I. erythromera (above, right), and I. henselii (below, right).
Acknowledgements
We thank Center for Scientific Computing (NCC/GridUNESP) of the UNESP and Cyberinfrastructure for Phylogenetic Research (CIPRES) for computer resources; Centro de Estudos de Insetos Sociais (CEIS; São Paulo State University [UNESP]) for allowing us the use of the molecular laboratory. A. Brunetti, C. Cassini, T. Silva Soares, D. Baêta, A. F. Sabbag, and J. S. Parreiras gave field assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Barrow L. N., A. R. Lemmon, E. Moriarty Lemmon. 2018. Targeted sampling and target capture: Assessing phylogeographic concordance with genome-wide data. Syst. Biol. Advance Access Syy021. (Accessed 21 March 2018).
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Bernt M, Braband A, Schierwater B, Stadler PF. 2013. Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol. 69:328–338.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Canedo C, Haddad CFB. 2012. Phylogenetic relationships within anuran clade Terrarana, with emphasis on the placement of Brazilian Atlantic rainforest frogs genus Ischnocnema (Anura: Brachycephalidae). Mol Phylogenet Evol. 65:610–620.
- Frost DR. 2018. Amphibian species of the world: an online reference. New York, USA: American Museum of Natural History.
- Gehara M, Canedo C, Haddad CFB, Vences M. 2013. From widespread to microendemic: molecular and acoustic analyses show that Ischnocnema guentheri (Amphibia: Brachycephalidae) is endemic to Rio de Janeiro, Brazil. Conserv Genet. 14:973–982.
- Goloboff PA, Catalano SA. 2016. TNT version 1.5, including a full implementation of phylogenetic morphometrics. Cladistics. 32:221–238.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucl Acids Res. 41:e129.
- Heinicke M., Lemmon A.R., Moriarty Lemmon E., McGrath K., Blair Hedges S.. 2018. Phylogenomic support for evolutionary relationships of New World direct-developing frogs (Anura: Terraranae). Mol. Phylogenet. Evol. 118:145–155.
- Heyer WR. 1984. Variation, systematics, and zoogeography of Eleutherodactylus guentheri and closely related species (Amphibia: Anura: Leptodactylidae). Smith Contr Zool. 402:1.
- Jiménez de la Espada M. 1870. Fauna neotropicalis species quaedam nondum cognitae. J Sci Math Phys Nat. 3:57–65.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kurabayashi A, Sumida M. 2013. Afrobatrachian mitochondrial genomes: genome reorganization, gene rearrangement mechanisms, and evolutionary trends of duplicated and rearranged genes. BMC Genomics. 14:633.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. Partitionfinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34:772–773.
- Lemmon AR, Emme SA, Lemmon EM. 2012. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst Biol. 61:727–744.
- Lutz A. 1925. Batraciens du Brésil. Comptes Rendus Mémoires Hebd des Séances la Société Biol des Ses Fil. 22:211–214.
- Padial JM, Grant T, Frost DR. 2014. Molecular systematics of terraranas (Anura: Brachycephaloidea) with an assessment of the effects of alignment and optimality criteria. Zootaxa. 3825:1.
- Peters WCH. 1870. Über neue Amphien (Hemidactylus, Urosaura, Tropdolepisma, Geophis, Uriechis, Scaphiophis, Hoplocephalus, Rana, Entomoglossus, Cystignathus, Hylodes, Arthroleptis, Phyllobates, Cophomantis) des Königlich Zoologisch Museum. Monatsberichte der Königlichen Preuss Akad des Wissenschaften Zu Berlin. 1870:641–652.
- Prum RO, Berv JS, Dornburg A, Field DJ, Townsend JP, Lemmon EM, Lemmon AR. 2015. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 526:569–573.
- Reinhardt JT, Lütken CF. 1862(1861). Bidrag til Kundskab om Brasiliens Padder og Krybdyr. Förste Afdeling: Padderne og Öglerne. Vidensk Meddelelser Fra Dansk Naturhistorisk Foren i Kjøbenhavn. 2:143–242.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Ruane S, Raxworthy CJ, Lemmon AR, Lemmon EM, Burbrink FT. 2015. Comparing species tree estimation with large anchored phylogenomic and small Sanger-sequenced molecular datasets: an empirical study on Malagasy pseudoxyrhophiine snakes. BMC Evol Biol. 15:1–14.
- Sabaj MH. 2016. Standard symbolic codes for institutional resource collections in herpetology and ichthyology: an online reference. Version 6.5 [accessed 2016 Aug 16]. http://www.asih.org/, American Society of Ichthyologists and Herpetologists 5:802–832. http://www.asih.org/resources, http://www.webcitation.org/6lkBdh0EO [accessed 2016 Nov 3].
- San Mauro D, Gower DJ, Zardoya R, Wilkinson M. 2006. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol Biol Evol. 23:227–234.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Steindachner F. 1864. Batrachologische Mittheilungen. Verhandlungen des Zool. Vereins Wien. 14:239–288.
- Taucce PPG, Canedo C, Parreiras JS, Drummond LO, Nogueira-Costa P, Haddad CFB. 2018. Molecular phylogeny of Ischnocnema (Anura: Brachycephalidae) with the redefinition of its series and the desctiption of two new species. Molecular Phylogenetics and Evolution. doi:10.1016/j.ympev.2018.06.042
- Tucker DB, Colli GR, Giugliano LG, Hedges SB, Hendry CR, Lemmon EM, Lemmon AR, Sites JW, Pyron RA. 2016. Methodological congruence in phylogenomic analyses with morphological support for teiid lizards (Sauria: Teiidae). Mol Phylogenet Evol. 103:75–84.
- Zhang P, Liang D, Mao RL, Hillis DM, Wake DB, Cannatella DC. 2013. Efficient sequencing of anuran mtDNAs and a mitogenomic exploration of the phylogeny and evolution of frogs. Mol Biol Evol. 30:1899–1915.