
Abstract
The Coryphaenidae family comprises of single genus, Coryphaena, which includes two species: Coryphaena hippurus and Coryphaena equiselis. In this study, we described the complete mitochondrial genome of the two species in Coryphaena. The assembled mitogenome of C. hippurus and C. equiselis consists of 16731 bp and 16690 bp, respectively. Two mitogenomes contain the typical gene complement including 13 protein-coding genes, 22 transfer RNAs, 2 ribosomal RNA genes and a non-coding D-loop. The longest protein-coding genes of these species was ND5, whereas the shortest ATP8. The length of D-loop is 1168 bp (C. hippurus) and 1206 bp (C. equiselis).
The Coryphaenidae family is comprised of single genus, Coryphaena, which only includes two species: Coryphaena hippurus and Coryphaena equiselis (Collette Citation1999). The dolphinfish is widely distributed in tropical and subtropical waters throughout the Atlantic, Indian and Pacific Oceans including the Mediterranean Sea (Gibbs and Collette Citation1959). Here, we sequenced and annotate mitogenome of two species, C. hippurus and C. equiselis in Coryphaena from South China Sea to provide molecular information for genetically understanding of dolphin fishes. The specimens of C. hippurus and C. equiselis were collected from two locations in the South China Sea (21°25′N, 117°02′E and 20°28′N, 120°03′E) in September 2017. Whole genomic DNA was extracted from muscle tissue of one specimen of each species using TIANamp Marine Animals DNA Kit (TIANGEN, China). The concentration for use as a PCR template was adjusted to an A260 of about 0.05 to 0.2. The collected specimens and extracted DNA were stored in Guangdong Provincial Key Laboratory of Fishery Ecology and Environment. The complete mitochondrial genomes were sequenced using PCR primers designed from highly conserved regions of transfer RNA (tRNA) sequences of congeneric species (Diaz-Jaimes et al. Citation2015) with additional specific primers designed as required from sequences already obtained. Long-PCR amplifications were performed by thermo-cycling using five pairs of primers and PCR amplicons were subjected to build up genomic library and pair-end sequencing by MiSeq. The COI sequence was used as reference seeds for iterative assembly by MITObim v.1.8 (Hahn et al. Citation2013). SeqMan v.7.1.0 was used for the mitogenome assembly and annotation (Swindell and Plasterer Citation1997). Transfer RNA genes were predicted using online software tRNAScan-SE 1.21 (Lowe and Eddy Citation1997). All the Protein coding gene (PCGs) are aligned independently, then concatenated to be applied for phylogenetic reconstruction with other Carangiformes in MrBayes v 3.12 (Ronquist and Huelsenbeck Citation2003) using relaxed clock model.
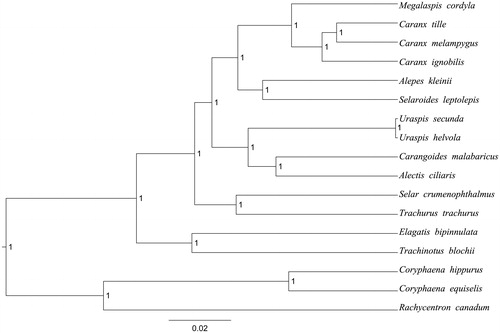
Complete mitogenomes of C. hippurus and C. equiselis consist of 16731 bp (GeneBank: MH576915) and 16690 bp (GeneBank: MH576916), respectively. These mitogenomes contain the typical gene complement including 13 protein-coding gene, 22 transfer RNA genes and 2 rRNA genes (12S rRNA and 16S rRNA) and one A + T-rich region which could also be termed as control region. The overall base composition of the whole mitochondrial genome of C. hippurus was A (26.69%), T (27.20%), G (15.61%) and C (23.88%) with an AT bias of 53.89%. The overall base composition of C. equiselis was A (27.11%), T (28.55%), G (16.71%), and C (24.32%) with an AT bias of 55.66%. In the two species, the ATG initiation codon are used in all protein-coding genes except COX1 (GTG) and ATP8 (ATT), the stop codons of all the 13 protein-coding genes were complete. Meanwhile, the longest protein-coding genes of these species was ND5 (1818 bp in C. hippurus; 1839 in C. equiselis), whereas the shortest ATP8 (165 bp in C. hippurus; 168 in C. equiselis). lrRNA and srRNA genes are 1719 bp and 946 bp in length in C. hippurus, 1721 bp and 946 bp in length in C. equiselis, and the length of D-loop is 1168 bp (C. hippurus) and 1206 bp (C. equiselis). All the 22 typical tRNAs possess a complete clover leaf secondary structure, ranging from 66 bp to 75 bp. The Bayesian inference phylogenetic tree showed that the two species firstly grouped together (). We have the confidence to construct phylogenetic trees, based on the complete the mitochondrial genomes, but the evolution history of dolphinfishes still needs future research to be clearly resolved.
Figure 1. The Bayesian inference phylogenetic tree for Carangiformes based on mitochondrial PCGs and rRNAs concatenated dataset. The gene’s accession numbers for tree construction are listed as follows: Elagatis bipinnulata (KT824759), Uraspis helvola (KM978993), Uraspis secunda (KT819204), Megalaspis cordyla (KM522836), Alepes kleinii (KF728081), Carangoides malabaricus (KJ174514), Selaroides leptolepis (KM522839), Trachurus trachurus (AB108498), Trachinotus blochii (KJ184305), Caranx ignobilis (KF649842), Caranx melampygus (KF649843), Caranx tille (KT805946), Alectis ciliaris (KM522837), Selar crumenophthalmus (KJ148633), Rachycentron canadum (FJ154956).
Disclosure statement
None of the co-authors has any conflict of interest to declare. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Collette BB, (1999). Coryphaenidae. Dolphinfishes, “dolphins”. In CKE, Niem VH. editors. FAO species identification guide for fishery purposes. The living marine resources of the Western Central Pacific. Volume 4. Bony fishes part 2 (Mugilidae to Carangidae). Rome: FAO. p 2656–2658.
- Diaz-Jaimes P, Bayona-Vasquez N, Hinojosa-Alvarez S, Uribe-Alcocer M, Marcet-Houben M. 2015. The complete mitogenome of the common dolphinfish (Coryphaena hippurus). Mitochondrial DNA. 26:959–960.
- Gibbs RH, Jr, Collette BB. 1959. On the identification, distribution,and biology of the dolphins, Coryphaena hippurus and C. equiselis. Bull Mar Sci Gulf Caribbean. 9:117–152.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19:1572–1574.
- Swindell SR, Plasterer TN. 1997. Seqman, contig assembly. In: Swindell SR, editor. Sequence data analysis guidebook. Totowa (NJ): Springer; p. 75–89.