
Abstract
Sweet cherry (Cerasus avium (L.) Moench) belonging to family Rosaceae, is an important economical fruit crop worldwide. In this study, the complete chloroplast (cp) genome of sweet cherry was generated by De novo assembly with low coverage whole-genome sequencing data. The genome size was 157,987 bp in length consisting of a typical quadripartite structure; a large single-copy region (LSC, 85,975 bp), a small single-copy region (SSC, 19,121 bp) and a pair of inverted repeat regions (IRs, 26,445 bp each). A total of 115 genes were predicted including 82 protein-coding genes, 29 tRNA genes and four rRNA genes. Phylogenetic analysis based on 12 reported complete chloroplast genome indicated the monophyly of the genus Creasus including newly sequenced C. avium, which is conform to the traditional classification.
Sweet cherry (Cerasus avium (L.) Moench) is one of the four cultivated cherry species worldwide. Its production has increased over 30% during the last two decades (Calle et al. Citation2018). It is reported that cultivated sweet cherry was domesticated from its wild ancestor ‘Mazzard’ in 4000–5000 years ago and also revealed one domestication event (Janick Citation2005; Meyer and Purugganan Citation2013). Despite the wide distribution and ample species of genus Cerasus, the genetic relationship of C. avium relative to other cherries has not been well established in the previous phylogenetic analyses using several nuclear and chloroplast markers (Pervaiz et al. Citation2015), especially for Chinese cherry (Cerasus pseudocerasus) and sweet cherry (C. avium) (Shi et al. Citation2013). Here, we generated the complete chloroplast genome sequence of C. avium to elucidate the phylogenetic relationship between C. avium and other cherries.
Total genomic DNA of ‘Mazzard’ was isolated from fresh leaves sampled from national fruit germplasm repository (kindly provided by Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences) using modified CTAB protocol. Voucher specimen .was deposited in Sichuan Agricultural University. Illumina paired-end (PE) library with 500-bp insert size was constructed and sequenced using Illumina HiSeq 2500 platform (Illumina, San Diego, CA). After quality trimming, a total of 4.23 Gb clean PE reads (Phred scores >20) were assembled using SOAPdenovo software (Li et al. Citation2009). Contigs were ordered and merged with the chloroplast sequence of Cerasus persica (NC_04697). Genome annotation was performed with Dual Organellar Geno Me Annotator (DOGMA) (Wyman et al. Citation2004) (http://dogma.ccbb.utexas.edu/)
The circular DNA of C. avium was 157,987 bp in length consisting of four regions; large single copy region (LSC) of 85,975 bp, small single copy region (SSC) of 19,121 bp, and a pair of inverted repeat regions (IRs) of each 26,445 bp. The overall GC contents were 35.72%. A total 115 unique coding regions were predicted, comprising 82 protein-coding genes, 29 tRNA genes, and four rRNA genes. Among all unique genes, nine genes contain one intron, two genes (ycf3 and clpP) with two introns. All the coding regions accounted for 57.36% of the whole genome. The genome was deposited in GenBank (MH_756631).
Similarity complete chloroplast genome sequence of other 12 Rosales species (Pyrus pyrifolia NC_015996 and Pyrus spinosa NC_023130 as outgroups) were aligned using MAFFT 5 (Katoh et al. Citation2005). Maximum-likelihood (ML) analysis was implemented in RAxML v8.2.4 (Stamatakis Citation2006). Maximum parsimony (MP) and neighbour-joining (NJ) analysis were performed using MEGA 6.0 (Tamura et al. Citation2013) (http://www.megasoftware.net/).
Figure 1. Phylogenetic tree of Cerasus avium with other 13 species belonging to Prunoideae, Rosaceae family. Tree was inferred from the complete chloroplast genome sequences using the ML method with a GTR model, MP method, and NJ method with a K-2P model. Only the framework of the ML tree was presented. Numbers in the nodes were the bootstrap values from 1000 replicates with an arrangement of ML/MP/NJ methods. Symbol (I, II, III) in the nodes represent three clades in subfamily Prunoideae.
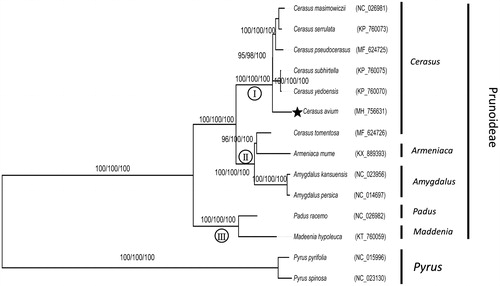
Phylogenetic analysis revealed three clades in subfamily Prunoideae (). C. avium was nested within genus Cerasus, which was a sister to other five Creasus species. C. tomentosa was nested within genus Armeniaca, and a sister to genus Amygdalus formed another clade. Genus Padus and Maddenia composed the third clade. This result was congruent with previous studies by other molecular markers (Wen et al. Citation2008; Chin et al. Citation2010; Chen et al. Citation2018).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Additional information
Funding
Related Research Data
References
- Calle A, Cai L, Iezzoni A, Wünsch A. 2018. High-density linkage maps constructed in sweet cherry (Prunus avium L.) using cross- and self-pollination populations reveal chromosomal homozygosity in inbred families and non-syntenic regions with the peach genome. Tree Genet Genome. 14:37.
- Chen T, Wang Y, Wang L, Chen Q, Zhang J, Tang HR, Wang XR. 2018. The complete chloroplast genome of Tomentosa cherry Prunus tomentosa (Prunoideae, Rosaceae). Mitochondrial DNA Part B. 3:672–673.
- Chin SW, Wen J, Johnson G, Potter D. 2010. Merging Maddenia with the morphologically diverse Prunus (Rosaceae). Bot J Linn Soc. 164:236–245.
- Janick J. 2005. The origins of fruits, fruit growing, and fruit breeding. Plant Breed Rev. 25:5–320.
- Katoh K, Kuma KI, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucl Acids Res. 33:511–518.
- Li R, Yu C, Li Y, Lam T-W, Yiu SM, Kristiansen K, Wang J. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 25:1966–1967.
- Meyer RS, Purugganan MD. 2013. Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet. 14:840–852.
- Pervaiz T, Sun X, Zhang YY, Tao R, Zhang JH, Fang JG. 2015. Association between chloroplast and mitochondrial DNA sequences in Chinese Prunus genotypes (Prunus persica, Prunus domestica, and Prunus avium). BMC Plant Bio. 15:4.
- Shi W, Wen J, Lutz S. 2013. Pollen morphology of the Maddenia clade of Prunus and its taxonomic and phylogenetic implications. J Syt Evol. 51:164–183.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22:2688–2690.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Bio Evol. 30:2725–2729.
- Wen J, Berggren ST, Lee CH, Ickert-Bond S, Yi TS, Yoo KO, Xie L, Shaw J, Potter D. 2008. Phylogenetic inferences in Prunus (Rosaceae) using chloroplast ndhF and nuclear ribosomal ITS sequences. J Syst Evol. 46:322–332.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.