
Abstract
We sequenced and assembled four whole mitogenome sequences of the bottom-dwelling fish yellowtail stargazer Uranoscopus cognatus isolated from East Peninsular Malaysia and West Coast of Thailand. The final partitioned nucleotide alignment consists of 14,098 bp and supports the monophyly of the genus Uranoscopus. Contrary to various mitogenome reports, we also made available the raw Illumina sequencing reads that can be further mined for microsatellite markers and repetitive nuclear genes.
Despite the recognition of Peninsular Malaysia as one of the world’s biodiversity hotspots, molecular resources of fishes in this tropic region are still relatively scarce with only one whole genome (Austin et al. Citation2015) and a few mitogenomes reported to date (Yue et al. Citation2006; Norfatimah et al. Citation2014; Gan et al. Citation2017). Recent advances in sequencing technology and software algorithms have enabled the rapid sequencing, assembly and annotation of whole mitogenome at a fraction of the cost and time of the conventional approach (Gan et al. Citation2014; Tan et al. Citation2015) thus creating an unprecedented opportunity for local researchers to perform a comprehensive molecular assessment of fishes in Malaysia. The Yellowtail stargazer (Uranoscopus cognatus) is a bottom-dwelling fish that buries itself in the sand. It exhibits a broad geographical distribution ranging from Thailand to North Australia, with its type locality designated as the Sea of Penang, Malaysia (Cantor Citation1850). Despite its prevalence, almost no molecular data are available for this species except for a 168 bp fragment of 12S rRNA reported for the voucher specimen KAUM:I:60990 (Accession number: LC037148). To contribute more significantly to the molecular resources for this species, we performed shallow shotgun sequencing of four U. cognatus individuals collected from the South China Sea near East Peninsular Malaysia (Isolates UMTGen1318 and UMTGen1321) and the Andaman Sea near Thailand (Isolates THNHM_F0014811 and THNHM_F0014643). Contrary to various mitogenome reports, in addition to the whole mitogenome sequences, we also made available the raw sequencing data that can be mined for microsatellites and phylogenetically informative nuclear genes (Straub et al. Citation2012; Thai et al. Citation2016; Grandjean et al. Citation2017).
DNA was extracted from the fin clip using salt extraction method (Miller et al. Citation1988). Approximately 100 ng of DNA was sheared to 300bp using QSonica Q800R2 (Qsonica LLT, Newton, CT) and processed with the NEBNextUltra DNA library preparation kit (New England Biolabs, Ipswich, MA). Shallow shotgun sequencing was performed on a Novaseq6000 (2 × 150 bp run configuration) located at the Deakin Genomics Centre. Mitogenome assembly used Novoplasty v2.7.2 (Dierckxsens et al. Citation2017) with the 12S rRNA sequence of U. cognatus chosen as the “seed sequence”. The assembled mitogenomes were annotated using Mitoannotator (Iwasaki et al. Citation2013). Thirteen protein-coding genes from the annotated mitogenomes of U. cognatus and its related species were extracted, partitioned by codon position and aligned using TranslatorX (Abascal et al. Citation2010) while the mitochondrial ribosomal RNAs were separately aligned using Muscle (Edgar Citation2004). Calculation of the best-fit partitioning scheme from the 41 nucleotide alignments (13 PCGs ×3 codon positions and 2 rRNA alignments) and maximum likelihood tree construction(1000 ultrafast bootstrap approximation) used IQ-Tree version 1.5.6 (Nguyen et al. Citation2015).
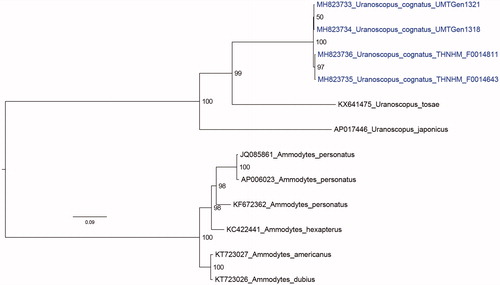
On average 148 million paired-end reads were generated per sample (minimum: 140 million;maximum: 156 million). In all four samples, the whole mitogenome was assembled except for the repetitive control region that could not be fully spanned by Illumina short reads. The initial 41 partitions were optimally merged into 4 partitions with the best-fit partitioning scheme as follows (number after gene name indicates codon position): Partition1 = ATP6-1,ND2-1,ND4-1,ND5-1,ND4L-1,COB-1,ND3-1,ND1-1,ATP8-1,ATP8-2, rrnL,rrnS,COX1-1,COX2-1,COX3-1; Partition2 = ATP6-2,NAD4-2,ND3-2,ND6-2,-ND4-2,ND2-2,ND5-2,COX1-2,COB-2,COX3-2,COX-2,ND1-2; Partition3 = ATP6-3,ND3-3,COB-3,COX3-3, NAD4L-3,NAD1-3,NAD2-3,NAD4-3,NAD5-3,ATP8-3,COX1-3; Partition4 = NAD6-1,NAD6-3. The monophyly of the genus Uranoscous is maximally supported by maximum likelihood analysis (). The pairwise nucleotide identity between individuals ranges from 98.999% to 99.827%. The U. cognatus samples formed a maximally-supported monophyletic group within the Uranoscopus clade () although clustering between samples collected from the Malaysia was not strongly supported. The mitogenome sequences have been submitted to NCBI under the accession numbers MH823733-MH823736. In addition, raw sequencing reads have been deposited in the SRA under the experiment number SRP159695.
Figure 1. Maximum likelihood tree depicting the evolutionary relationships of Uranoscopus cognatus and related species based on 13 mitochondrial protein-coding genes and 2 ribosomal RNAs (total alignment length of 14,098 bp) rooted with members of the genus Ammodytes as the outgroup. Mitogenomes reported in this study were colored blue. Numbers at nodes indicate IqTree Ultrafast Bootstrap support values and branch lengths indicate the number of substitutions per site.
Acknowledgements
We thank SEAFDEC/MFRDMD, SEAFDEC TD Bangkok, Malaysia Department of Fisheries and LKIM for the samples from East Peninsular Malaysia. We thank Sahat Ratmuangkhwang (Andaman Coastal Research Station for Development, Kasetsart University) for his kind assistance in collecting the sample tissues from Thailand.
Disclosure statement
The authors report no conflicts of interest and are responsible for the content and writing of the manuscript.
Additional information
Funding
Related Research Data
References
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38:W7–13.
- Austin CM, Tan MH, Croft LJ, Hammer MP, Gan HM. 2015. Whole genome sequencing of the Asian arowana (Scleropages formosus) provides insights into the evolution of ray-finned fishes. Genome Biol Evol. 7:2885–2895.
- Cantor TE. 1850. Catalogue of Malayan fishes. J Asiatic Soc Bengal. 18:983–1443.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5:113.
- Gan HM, Amornsakun T, Tan MP. 2017. The complete mitochondrial genome of the snakeskin gourami, Trichopodus pectoralis (Regan 1910) (Teleostei: Osphronemidae). Mitochondrial DNA B. 2:148–149.
- Gan HM, Schultz MB, Austin CM. 2014. Integrated shotgun sequencing and bioinformatics pipeline allows ultra-fast mitogenome recovery and confirms substantial gene rearrangements in Australian freshwater crayfishes. BMC Evol Biol. 14:19.
- Grandjean F, Tan MH, Gan HM, Lee YP, Kawai T, Distefano RJ, Blaha M, Roles AJ, Austin CM. 2017. Rapid recovery of nuclear and mitochondrial genes by genome skimming from Northern Hemisphere freshwater crayfish. Zool Scripta. 46:718–728.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30:2531–2540.
- Miller SA, Dykes DD, Polesky HF. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Norfatimah M, Teh L, Salleh M, Isa MM, SitiAzizah M. 2014. Complete mitochondrial genome of Malaysian Mahseer (Tor tambroides). Gene. 548:263–269.
- Straub SCK, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. 2012. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 99:349–364.
- Tan MH, Gan HM, Schultz MB, Austin CM. 2015. MitoPhAST, a new automated mitogenomic phylogeny tool in the post-genomic era with a case study of 89 decapod mitogenomes including eight new freshwater crayfish mitogenomes. Mol. Phylogenet. Evol. 85:180–188.
- Thai BT, Tan MH, Lee YP, Gan HM, Tran TT, Austin CM. 2016. Characterisation of 12 microsatellite loci in the Vietnamese commercial clam Lutraria rhynchaena Jonas 1844 (Heterodonta: Bivalvia: Mactridae) through next-generation sequencing. Mol Biol Rep. 43:391–396.
- Yue GH, Liew WC, Orban L. 2006. The complete mitochondrial genome of a basal teleost, the Asian arowana (Scleropages formosus, Osteoglossidae). BMC Genomics. 7:242.