
Abstract
Casuarina glauca, commonly known as swamp oak, belongs to Casuarinaceae. Herein, we reported the complete chloroplast genome of C. glauca. To our knowledge, this is the first reported complete chloroplast genome of Casuarinaceae species. The complete chloroplast genome of C. glauca is 155,669 bp in length with a typical quadripartite structure, consisting of a large single-copy region (LSC, 85,823 bp), a single-copy region (SSC, 25,694 bp) and a pair of inverted repeats (IRs, 18,461 bp). There are 130 genes annotated, including 85 protein-coding genes, eight ribosomal RNA genes, and 36 transfer RNA genes. To investigate the evolution status of C. glauca, as well as Casuarinaceae, we constructed a phylogenetic tree with C. glauca and other 16 species based on their complete chloroplast genomes. According to the phylogenetic topologies, C. glauca was closely related to Betulaceae.
Casuarina glauca, also known as swamp oak, has been widely planted as a street tree (Cheng Citation1982). C. glauca belongs to Casuarinaceae, members of which are a valuable resource of high-quality wood and are essential for the recovery of coastal zone ecosystem. The de novo genome C. glauca has been reported in a recent study (Griesmann et al. Citation2018). However, the analysis based on its complete chloroplast genome is still lack. In this study, we reported the complete chloroplast genome of C. glauca, and explored the evolution status of C. glauca, as well as Casuarinaceae, based on a phylogenetic tree.
In this study, ∼11G high-quality Illumina reads of C. glauca were downloaded from NCBI (SRR5311007). The sample of the published sequencing data was collected in Australia and was stored in Australian Tree Seed Center, CSIRO, Australia with the accession number N°15932 (Griesmann et al. Citation2018). We extracted the chloroplast-related reads by software Bwa (Li Citation2013) and Samtools (Li et al. Citation2009). Then, we used the software NOVOPlasty v2.5.9 (Dierckxsens et al. Citation2017), GapCloser (Luo et al. Citation2012), and Genious version 11.1.14 (Kearse et al. Citation2012) to assemble the complete chloroplast genome of C. glauca. The annotation was carried out with the software Plann (Huang and Cronk Citation2015) and Sequin (NCBI website). Finally, the annotated complete chloroplast genome of C. glauca was submitted to GenBank under the accession number of MH824417.
In total, the size of complete chloroplast genome of C. glauca is 155,669 bp. It has a typical quadripartite structure, including a large single-copy region (LSC, 85,823 bp), a small single-copy region (SSC, 25,694 bp), and a pair of inverted repeats (IRA and IRB, 18,461 bp). The overall GC content of the genome is 36.37%. There are 130 genes annotated in the complete chloroplast genome of C. glauca, including eight ribosomal RNA (rRNA) genes, 36 transfer RNA (tRNA) genes and 85 protein-coding genes (PCGs). Most of these are single-copy genes, while 17 genes (six PCGs genes, four rRNA genes, and seven tRNA genes) located in the IR regions.
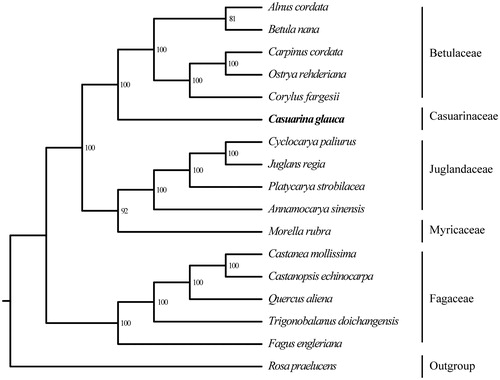
To explore the evolution status of C. glauca, as well as Casuarinaceae, we inferred the phylogenetic relationships based on the complete chloroplast genomes of 17 species, including C. glauca, 15 Fagales species, and the outgroup Rosa praelucens. The alignment was carried out by software MAFFT (Katoh and Standley Citation2013). And the phylogenetic tree was generated by the software RaxML (Stamatakis Citation2014) with 100 bootstrap replicates (). Consistent to the previous studies (Li et al. Citation2004; Magallón et al. Citation2015), C. glauca is closely related to Betulaceae. In summary, this newly characterized complete chloroplast genome could provide essential data for a better understanding of C. glauca, and for further studies on the evolution of Casuarinaceae.
Figure 1. Maximum likelihood (ML) tree based on the complete chloroplast genome sequences of Casuarina glauca and other 16 species. Numbers in the nodes are the bootstrap values from 100 replicates. Their accession numbers are as follows: Alnus cordata: NC_036751; Betula nana: NC_033978; Carpinus cordata: NC_036723; Ostrya rehderiana: NC_028349; Corylus fargesii: KX822767; Cyclocarya paliurus: NC_034315; Juglans regia: NC_028617; Platycarya strobilacea: NC_035413; Annamocarya sinensis: KX703001; Morella rubra: NC_035006; Castanea mollissima: NC_014674; Castanopsis echinocarpa: NC_023801; Quercus aliena: NC_026790; Trigonobalanus doichangensis: NC_023959; Fagus engleriana: NC_036929; Rosa praelucens: NC_037492.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Cheng Y. 1982. Flora of China. Science Press. 20(1):3.
- Dierckxsens N, Mardulyn P, Smits G. 2017. Novoplasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 4:e18.
- Griesmann M, Chang Y, Liu X, Song Y, Haberer G, Crook MB. 2018. Phylogenomics reveals multiple losses of nitrogen-fixing root nodule symbiosis. Science. 361:eaat1743.
- Huang DI, Cronk QCB. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Li RQ, Chen ZD, Lu AM, Soltis DE, Solt PS, Manos PS. 2004. Phylogenetic relationships in Fagales based on DNA sequences from three genomes. Int J Plant Sci. 165:311–324.
- Li H. 2013. Aligning sequence reads. Clone Sequences and Assembly Contigs with Bwa-Mem. 1303. arXiv preprint arXiv:1303.3997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. 2012. Soapdenovo2: an empirically improved memory-efficient short-read de novo, assembler. GigaScience. 1:1–6.
- Magallón S, Gómez-Acevedo S, Sánchez-Reyes LL, Hernández-Hernández T. 2015. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist. 207:437–453.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.