
Abstract
The Madagascar mother-of-pearl (Salamis anteva) is a leaf-mimicking butterfly endemic to forests in southern Madagascar. Genome skimming by Illumina sequencing permitted assembly of a complete S. anteva circular mitogenome of 15,201 bp consisting of 80.6% AT nucleotides and with an arrangement of 22 tRNAs, 13 protein-coding genes, two rRNAs and a control region common to all butterflies. The mitogenome includes a COX1 gene with an atypical CGA start codon and four genes exhibiting incomplete stop codons. Phylogenetic reconstruction places S. antvea as sister to Yoma sabina within a monophyletic nymphalid tribe Junoniini. This is consistent with previous molecular phylogenetic hypotheses.
The Madagascar mother-of-pearl butterfly, Salamis anteva (Lepidoptera: Nymphalidae), is a leaf-mimicing endemic species (Suzuki et al. Citation2014) that inhabits forested regions in the southern portions of Madagascar (Lees et al. Citation2003). It has been suggested as an indicator species for the health of these forested habitats (Kremen Citation1994). Here, we report the complete mitochondrial genome sequence of S. anteva from a specimen (ID: Sant2015.1) collected in Andasibe, Madagascar in March 2015 that has been pinned, spread, and deposited in the Wallis Roughley Museum of Entomology, University of Manitoba (voucher JBWM0379998).
DNA was prepared (McCullagh and Marcus Citation2015) and sequenced by Illumina MiSeq (San Diego, California) (Peters and Marcus Citation2017). The mitogenome of S. anteva (Genbank MH917707) was assembled by Geneious 10.1.2 from 7,876,900 paired 300 bp reads using a Junonia lemonias (Nymphalidae, KP941756) (McCullagh and Marcus Citation2015) reference mitogenome and annotated by comparison with J. lemonias and Precis andremiaja (Nymphalidae: MH917706) (Lalonde and Marcus Citation2018) mitogenomes. The S. anteva nuclear rRNA repeat (Genbank MH917709) was assembled and annotated using Meroptera pravella (MF073208) (Living Prairie Mitogenomics Consortium Citation2017), Samia cynthia ricini (Saturniidae, AF463459) (Wang et al. Citation2003) and Papilio xuthus (Papilionidae, AB674749) (Futahashi et al. Citation2012) reference sequences.
The Salamis anteva circular 15,201 bp mitogenome assembly was composed of 4040 paired reads with nucleotide composition: 40.5% A, 11.8% C, 7.6% G, and 39.9% T. The gene composition and order in the P. andremiaja mitogenome matches those of other butterflies (McCullagh and Marcus Citation2015). Precis andremiaja mitochondrial protein-coding genes use the start condons: ATG (COX2, ATP6, COX3, NAD4, NAD4L, CYTB, NAD1), ATT (NAD2, ATP8, NAD6), ATC (NAD3), ATA (NAD5), and CGA (COX1). The mitogenome contains three protein-coding genes (COX1, COX2, NAD4) with single-nucleotide (T) stop codons and one gene (NAD2) with a two-nucleotide (TA) stop codon completed by adding 3′ A residues post-transcriptionally. The NAD5 gene terminates with an unusual TAG stop codon. The mitochondrial transcription termination site in S. anteva is GTACTAAAT, while in most species in the tribe Junoniini the first and last positions of this binding site are adenine (A) bases. The locations and structures of tRNAs were determined using ARWEN v.1.2 (Laslett and Canback Citation2008). The mitochondrial rRNAs are typical of Lepidoptera (McCullagh and Marcus Citation2015) with characteristic cloverleaf secondary structures except for trnS (AGN) where a loop replaces the dihydrouridine arm.
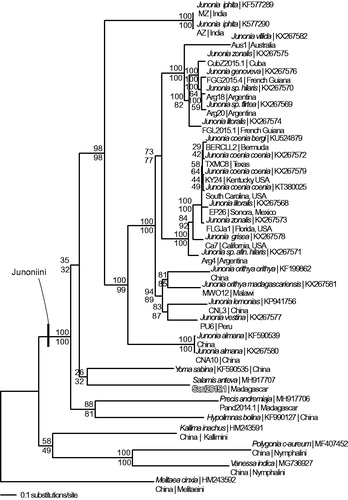
Phylogenetic reconstructions used complete mitogenomes from S. anteva, 25 representatives from tribe Junonini, and 4 outgroup species from other tribes within the Nymphalinae (McCullagh and Marcus Citation2015; Peters and Marcus Citation2016; Peters and Marcus Citation2017; Lalonde and Marcus Citation2018; McCullagh and Marcus Citation2018). Sequences were aligned in CLUSTAL Omega (Sievers et al. Citation2011) and analyzed by parsimony and maximum likelihood (model selected by jModeltest 2.1.7 (Darriba et al. Citation2012) and likelihood ratio test (Huelsenbeck and Rannala Citation1997)) algorithms in PAUP* 4.0b8/4.0d78 (Swofford Citation2002) (). Phylogenetic analysis places S. anteva as the sister-taxon to Yoma sabina within tribe Junoniini which is congruent with previous molecular phylogenetic analyses (Wahlberg et al. Citation2005; Kodandaramaiah and Wahlberg Citation2007).
Figure 1. Maximum likelihood phylogeny (GTR + G model, Gamma =0.1930, likelihood score 75924.15364) of Salamis anteva, 25 additional mitogenomes from tribe Junonini, and 4 outgroup species from other tribes in subfamily Nymphalinae based on 1 million random addition heuristic search replicates (with tree bisection and reconnection). One million maximum parsimony heuristic search replicates produced 24 equally most parsimonious trees (parsimony score 11461 steps) one of which is identical to the topology of the maximum likelihood tree. Numbers above each node are maximum likelihood bootstrap values and numbers below each node are maximum parsimony bootstrap values (each from 1 million random fast addition search replicates).
Acknowledgements
We thank Aleksandar Ilik and Debbie Tsuyuki (Children’s Hospital Research Institute of Manitoba Next Generation Sequencing Platform) for assistance with library preparation and sequencing.
Disclosure statement
The authors report no conflicts of interest and are solely responsible for this paper.
Additional information
Funding
Related Research Data
References
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772.
- Futahashi R, Shirataki H, Narita T, Mita K, Fujiwara H. 2012. Comprehensive microarray-based analysis for stage-specific larval camouflage pattern-associated genes in the swallowtail butterfly, Papilio xuthus. BMC Biology. 10:46.
- Huelsenbeck JP, Rannala B. 1997. Phylogenetic methods come of age: testing hypotheses in an evolutionary context. Science. 276:227–232.
- Kodandaramaiah U, Wahlberg N. 2007. Out-of-Africa origin and dispersal-mediated diversification of the butterfly genus Junonia (Nymphalidae: Nymphalinae). J Evolution Biol. 20:2181–2191.
- Kremen C. 1994. Biological inventory using target taxa: a case study of the butterflies of Madagascar. Ecol Appl. 4:407–422.
- Lalonde MML, Marcus JM. (2018). The complete mitochondrial genome of the Madagascar banded commodore butterfly Precis andremiaja (Insecta: Lepidoptera: Nymphalidae). Submitted to Mitochondrial DNA Part B: Resources. doi: 10.1080/23802359.2018.1541721.
- Laslett D, Canback B. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 24:172–175.
- Lees DC, Kremen C, Raharitsimba T. 2003. Classification, diversity, and endemism of the butterflies (Papilionoidea and Herperioidea): a revised species checklist. In: Goodman, SM and Benstead, JP, editors. The Natural History of Madagascar. Chicago and London: The University of Chicago Press; p. 762–792.
- Living Prairie Mitogenomics Consortium 2017. The complete mitochondrial genome of the lesser aspen webworm moth Meroptera pravella (Insecta: Lepidoptera: Pyralidae). Mitochondrial DNA B Resour. 2:344–346.
- McCullagh BS, Marcus JM. (2018). When barcodes go bad: Exploring the limits of DNA barcoding with complete Junonia butterfly mitochondrial genomes Submitted to Molecular Phylogenetics and Evolution: Manuscript #MPE_2017_9.
- McCullagh BS, Marcus JM. 2015. The complete mitochondrional genome of Lemon Pansy, Junonia lemonias (Lepidoptera: Nymphalidae: Nymphalinae). J Asia-Pacific Ent. 18:749–755.
- Peters MJ, Marcus JM. 2016. The complete mitochondrial genome of the Bermuda buckeye butterfly Junonia coenia bergi (Insecta: Lepidoptera: Nymphalidae). Mitochondrial DNA B Resour. 1:739–741.
- Peters MJ, Marcus JM. 2017. Taxonomy as a hypothesis: testing the status of the Bermuda buckeye butterfly Junonia coenia bergi (Lepidoptera: Nymphalidae). Syst Ent. 42:288–300.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. 2011. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Syst Biol. 7:539.
- Suzuki TK, Tomita S, Sezutsu H. 2014. Gradual and contigent evolutionary emergence of leaf mimicry in butterfly wing patterns. BMC Evolutionary Biology. 14:229.
- Swofford DL. 2002. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sunderland, Massachusetts, USA: Sinauer Associates.
- Wahlberg N, Brower AVZ, Nylin S. 2005. Phylogenetic relationships and historical biogeography of tribes and genera in the subfamily Nymphalinae (Lepidoptera: Nymphalidae). Biol J Linn Soc. 86:227–251.
- Wang SQ, Zhao MJ, Li TP. 2003. Complete sequence of the 10.3 kb silkworm Attacus ricini rDNA repeat, determination of the transcriptional initiation site and functional analysis of the intergenic spacer. DNA Sequence. 14:95–101.