
Abstract
Sonneratia apetala is a true mangrove distributed in South Asia, Southeast Asia and China. Here the whole chloroplast genome of S. apetala was assembled. The chloroplast genome was 153,052 bp in length, consisting of a large single copy (LSC) region of 87,210 bp, a small single copy (SSC) region of 18,026 bp, and two inverted repeat (IR) regions of 23,908 bp. It contained 131 genes, including 86 protein-coding genes, 37 tRNA genes and eight rRNA genes. The overall GC content was 37.3%. Phylogenetic analysis showed that S. apetala was sister to Trapa maximowiczii in Lythraceae.
Mangroves belong to different families or orders but have undergone genome-wide convergent evolution to adapt to coastal saline or brackish water (Xu et al. Citation2017). Sonneratia apetala Buch.-Ham. (Lythraceae) is a small to medium size columnar true mangrove, which is native to South Asia and Southeast Asia, such as India, Bangladesh, and Myanmar. Although this species is fast-growing, the seed viability is low. Due to habitat loss or extraction, as well as localized extinction of the species in some parts of south Asia, there has been an estimated 7% decline within the species range since 1980 (FAO Citation2007). Sonneratia apetala is listed in the IUCN Red List of Threatened Species (Kathiresan et al. Citation2010).
In this study, we reconstructed the complete chloroplast (cp) genome of S. apetala. The annotated cp genome has been deposited in GenBank with accession number MH986669. Total genome DNA was extracted from fresh leaves of an individual of S. apetala, collected from Hepu, Guangxi, China (N21°29′53″, E109°45′39″) and deposited at South China Botanical Garden Herbarium (ZhouLX13699), using a modified CTAB protocol (Doyle and Doyle Citation1987). Library construction and paired-end sequencing on Illumina HiSeq 2500 systems were performed by Beijing Genomics Institute (Shenzhen, China). Genome sequences were assembled in NOVOPlasty (Dierckxsens et al. Citation2017). The genes in the chloroplast genome were annotated using GeSeq (Tillich et al. Citation2017), with manual adjustment using Geneious ver. 11.0.2 (Kearse et al. Citation2012).
The complete cp genome of S. apetala is 153,052 bp in length, consisting of a large single copy (LSC) region of 87,210 bp, a small single copy (SSC) region of 18,026 bp, and two inverted repeat (IR) regions of 23,908 bp. The overall GC content was 37.3%, higher than those of LSC (35.3%) and SSC (31.0%) regions, but lower than those of IR regions (43.1%). The chloroplast genome of S. apetala contained 131 genes, including 86 protein-coding genes, 37 tRNA genes and eight rRNA genes. Among the protein-coding genes, eight genes contain a single intron while three genes possess two introns. Most genes occurred in single copy, while six protein coding genes, seven tRNA genes, and four rRNA genes in IR regions were duplicated.
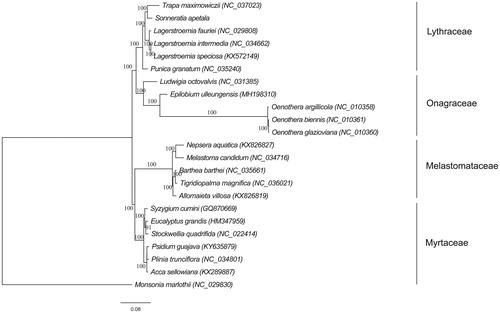
Phylogenomic analysis was performed using the complete chloroplast genomes from 22 species in the order Myrtales (including S. apetala in this study). Monsonia marlothii from Geraniales was used as outgroup species. The cp genome sequences were aligned using MAFFT (Katoh and Standley Citation2013). Phylogenetic tree was constructed with RAxML (Stamatakis Citation2014) implemented in Geneious, using the maximum-likelihood algorithm. The monophyly of the Lythraceae and the sister relationship between Sonneratia and Trapa were supported in this study (). The identified evolutionary relationships of Lythraceae, Onagraceae, Myrtaceae, and Melastomataceae were consistent with the Angiosperm Phylogeny Group classification (The Angiosperm Phylogeny Group, Citation2016). The chloroplast genome of S. apetala is of significance for its conservation and evolutionary studies.
Figure 1. Maximum-likelihood tree inferred from chloroplast genome sequences of 22 species from the order Myrtales. The bootstrap values were based on 1000 replicates.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- FAO. 2007. The World's Mangroves 1980-2005. FAO Forestry Paper 153. Forestry Department, Food and Agriculture Organization of the United Nations (FAO), Rome.
- Kathiresan K, Salmo III SG, Fernando ES, Peras JR, Sukardjo S, Miyagi T, Ellison J, Koedam NE, Wang Y, Primavera J, et al. 2010. Sonneratia apetala. The IUCN Red List of Threatened Species 2010. http://dx.doi.org/10.2305/IUCN.UK.2010-2.RLTS.T178841A7623713.en.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- The Angiosperm Phylogeny Group. 2016. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 181:1–20.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes . Nucleic Acids Res. 45:W6–W11.
- Xu S, He Z, Guo Z, Zhang Z, Wyckoff GJ, Greenberg A, Wu C, Shi S. 2017. Genome-wide convergence during evolution of mangroves from woody plants. Mol Biol Evol. 34:1008–1015.