
Abstract
Genetic innovation may be linked to evolutionary success, and indeed, the invasive round goby mitochondrial genome sequence carries two novel features not previously described in Benthophilinae. First, the round goby mitochondrial genome carries a rearrangement of the tRNA cluster Ile-Glu-Met. Second, the round goby mitochondrial genome features a 1250 bp non-coding sequence insertion downstream of the D-loop region. In this publication, we test where in the goby phylogeny the novel tRNA arrangement first arose and whether the sequence insertion is associated with invasive populations only or a genuine feature of the species. We sequence native and invasive populations in Europe and North America, and show that all round gobies carry the sequence insertion. By sequencing the tRNA cluster in selected Gobiidae, we show that the tRNA arrangement arose at the root of the Benthophilinae species radiation.
Introduction
Recently, two novel mitochondrial genome features were reported in the round goby (Adrian-Kalchhauser et al. Citation2017). The round goby is an invasive, small, benthic Ponto-Caspian fish species. It is native to the Black and Caspian Sea and their tributaries, and belongs to one of three tribes within the family of Benthophilinae (Neilson and Stepien Citation2009a). Many species of this family have colonized rivers and coasts outside the native range in Europe and North America (Kornis et al. Citation2012; Roche et al. Citation2013). However, the round goby Neogobius melanostomus is the most successful invader among them (Hirsch et al. 2015).
Genetic innovation may be linked to evolutionary success, and indeed, the round goby mitochondrial genome sequence carries two novel features. First, the round goby mitochondrial genome carries a rearrangement of the tRNA cluster between the ND1 and ND2 genes. In gobies generally, the cluster contains tRNA Isoleucine (forward orientation), tRNA Glutamine (reverse orientation) and tRNA Methionine (forward orientation), without any spacer sequence. In the round goby, the position of tRNA Isoleucine and tRNA Glutamine are swapped to yield the sequence Gln (rev) Ile (fw) Met (fw), and tRNA genes are separated by up to 42 nucleotides of noncoding spacer sequence. Second, the round goby mitochondrial genome features a 1250 bp non-coding sequence insertion downstream of the D-loop region. The insert bears only minimal similarities to D-loop repeats or any known sequence, and is flanked on both sides by potentially functional genes for tRNA Phenylalanine.
In this publication, we trace the phylogenetic origin of these two novel features. We test (a) whether the novel tRNA arrangement first arose in the round goby, or originated earlier in the goby phylogeny, and (b) whether the sequence insertion is a universal feature present in all round gobies or an anomaly associated with certain invasive populations only. To locate the origin of the tRNA re-arrangement, we compared existing and newly sequenced tRNA cluster arrangements in 10 gobiid species (5 members of the Benthophilinae, and 5 members of other gobiid families). To determine whether the non-coding insert is a genuine feature of the species Neogobius melanostomus or an anomaly associated with the Swiss invasive population, we analyzed a 7.5 kb region of the mitochondrial genome using Oxford Nanopore long read technology in individuals from the native region and from three globally distributed invaded sites.
Materials and methods
Origin of the round goby Gln-Ile-Met tRNA rearrangment
Representative goby species were chosen for analysis based on previous phylogenetic analyses (Neilson and Stepien Citation2009a) to cover major branches of the Benthophilinae and sister groups. Sequences of Ponticola kessleri, Odontobutis obscura and Gillichthys mirabilis were available at NCBI (Sequence accession numbers: NC_025638.1, KT438552.1 and NC_012906.1, respectively). Sequences of Neogobius fluviatilis, Babka gymnotrachelus, Proterorhinus semilunaris, Zosterisessor ophiocephalus and Gobius niger were generated de novo in this study.
Tissue samples were obtained from collaborators (see acknowledgements) and shipped in 100% EtOH. DNA was isolated with standard phenol-chloroform extraction and precipitation in the following way: tissues were lysed in ATL lysis buffer from the DNeasy Blood & Tissue kit from QIAGEN according to the manufacturer’s instruction. For each 25 mg of tissue, 180 µL lysis buffer and 20 µL Proteinase K were used. After lysis, residual debris was removed by centrifugation at 4 °C for 10 min at 13,000 g in an Eppendorf Centrifuge 5423R. Then, proteins were removed by two rounds of phenol-chloroform-isoamylalcohol extraction (25:24:1; Roth) and one round of chloroform extraction. DNA was precipitated by adding 1/10 volume of sodium acetate (3 M, pH 5.2), and 2.5 volumes of 100% ethanol, incubating at −80 °C for several hours, and centrifuging at 13,000 g for 30 min in an Eppendorf Centrifuge 5423R. DNA pellets were washed by centrifugation in the presence of 1 ml of 70% EtOH for 10 min and then dried and dissolved in 100 µL ddH2O. DNA concentrations were measured using a NanoDrop™ 2000 spectrophotometer.
A 1850 bp fragment containing the genes for tRNAs Gln, Ile and Met () was amplified using primers fragment1_fw_mtGenome (CCCGATTCCGATATGACCAAC) and SG015 (CCACAGGTAAAATGGCTGAG) for Babka gymnotrachelus and Proterorhinus semilunaris, and primers SG006 (ATGAGTGCGAGCCTCCTACC) and SG015 (see above) for Neogobius fluviatilis, Proterorhinus semilunaris, Zosterisessor ophiocephalus and Gobius niger. The PCR mix was as follows: 14.4 µL ddH2O, 2 µL 10× Buffer + MgCl2, 1.6 µL 2.5 mM dNTPs, 1 µL DNA (1–100 ng), 0.2 µL FastStart Taq (Roche) and 0.4 µL 10 µM primer each. PCR cycling conditions were denaturation at 94 °C for 4 min, followed by 35 cycles of amplification with 94 °C for 30 sec, 50 °C for 30 sec and 72 °C for 2 min, followed by an elongation step at 72 °C for 10 min, and a final 4 °C step. Amplicon size was controlled by agarose gel electrophoresis.
PCR products were then cloned for sequencing. About 1 µL of fresh PCR product was directly used in TA-cloning reactions (TOPO® TA Cloning kit for sequencing, Thermo Fisher Scientific) using 0.5 µL TOPO®vector and incubation times of more than 30 min at RT. The TA cloning reactions were transformed by heat shock into competent DH5α bacteria prepared in-house. Candidate colonies were mini-prepped using the ZR Plasmid Miniprep Classic kit (Zymo Research) and sequenced at Microsynth with the company’s M13F and M13R primers. Sequences were analysed and inspected using ApE (A plasmid Editor, freely available at http://jorgensen.biology.utah.edu/wayned/ape/).
Origin of the large round goby non-coding mitochondrial genome insertion
Round goby samples were obtained from the native region and from the invasive range in North America and from the European invasive range from collaborators (see acknowledgements). Samples were shipped as tissue samples in ethanol.
DNA isolations were done as described above. Then, a 7.5 kb fragment of the mitochondrial genome spanning Cytochrome b, the control region, and the region coding for 12S and 16S ribosomal RNA () was amplified using the LongAmp®Taq2xMastermix (New England Biolabs) with forward primer SG082 (CCACCAACCCCAACAATAAG) and reverse primer SG083 (AAGCATAGTCAAGGGGAGGAG) according to the manufacturer’s instructions. PCR cycling conditions were 94 °C for 30 sec, followed by 35 cycles of amplification with 94 °C for 30 sec, 62.5 °C for 1 min, 65 °C for 7 min, followed by an elongation step at 65 °C for 10 min and a final 4 °C step. Amplicon size was controlled by agarose gel electrophoresis.
Then, samples were prepared for long-range sequencing with MinION technology (Oxford Nanopore). In a first step, PCR reactions were cleaned using a 1:1 ratio of Agencourt AMPure XP beads (Beckman Coulter) and eluted with 50 µL of ddH2O. Then, samples were dA-tailed using the NEBNext dA-Tailing Module (New England Biolabs) according to the protocol ‘1D PCR barcoding (96) genomic DNA (SQK-LSK108)’ using 45 µL DNA, 7 µL buffer, 3 µL Ultra II End-prep enzyme, 5 µL ddH2O, and incubated for 5 min at 20 °C and 5 min at 65 °C in a PCR machine. Residual nucleotides were removed by cleaning with Agencourt AMPure XP beads (Beckman Coulter) as above. Half of the beads were eluted with 10 µL of the respective barcode adaptor from the PCR 96 barcoding Kit (Oxford Nanopore) (instead of water; to reduce sample volume and save enzyme in the next step), the other half was kept as backup. 10 µL of Blunt/TA Ligase Master Mix (New England Biolabs) was added and samples were incubated for 30 min at RT. Reactions were cleaned using a 0.4 ratio of Agencourt AMPure XP beads (Beckman Coulter) and eluted with 25 µL of ddH2O.
Barcoding-PCR was performed using the LongAmp®Taq2xMastermix and the corresponding barcoding PCR primer. PCR cycling conditions were 95 °C for 3 min, followed by 22–29 cycles of amplification with 95 °C for 15 sec, 62.5 °C for 15 sec, 65 °C for 7 min, followed by an elongation step at 65 °C for 10 min and a final 4 °C step. Most often, 22 cycles yielded sufficient amounts of product. If the band was faint, cycle numbers were increased stepwise.
For sequencing, 10 µL of each product were combined. A 0.4 ratio of Agencourt AMPure XP beads (Beckman Coulter) was used for clean-up. 45 µL of the combined sample were then used in a dA-tailing reaction as done previously, with 5 µL internal control DNA provided in the kit added as spike. The reaction was cleaned up using DNA-low binding tubes and a 1:1 ratio of Agencourt AMPure XP beads (Beckman Coulter). The sample was then mixed on a thermomixer at 22 °C for 5 min and eluted using 35 µL of ddH2O.
30 µL (846 ng) of DNA were used in the adaptor ligation reaction. 20 µL adaptor mix (Ligation sequencing kit 1D, Oxford Nanopore) and 50 µL Blunt/TA Master Mix were added and incubated for 30 min at RT. A 0.4 ratio of Agencourt AMPure XP beads (Beckman Coulter) was used for clean-up and the library was eluted with 20 µL of ABB buffer (Ligation sequencing kit 1D, Oxford Nanopore). Loading of the MinION device was done as described by the 1D PCR barcoding (96) genomic DNA protocol (Oxford Nanopore). Sequencing (flowcell FLO-MIN106) was performed at the Genetic Diversity Centre Zurich. ONT Albacore Sequencing Pipeline Software (version 2.1.7) was use to basecall, demultiplex and aligned the fast5 files. NanoPack (De Coster et al. Citation2018) was used to create statistical summaries. We used samtools (V1.8, https://github.com/samtools/samtools) to combine sam files of the same barcode, convert the sam files to bam files, and sort the bam files. The bam files were inspected and consensus sequences extracted with IGV (Robinson et al. Citation2011; Thorvaldsdottir et al. Citation2013).
Results and discussion
We compared the Ile-Glu-Met tRNA cluster in five Benthophilinae (Ponticola kessleri, Babka gymnotrachelus, Proterorhinus semilunaris, Neogobius melanostomus and Neogobius fluviatilis) and in five non-Benthophilinae goby species (Zosterisessor ophicephalus, Gobius niger, Odontobutis obscura, Gillichtys mirabilits and Pomatoschistus minutus) and found that all analysed Benthophilinae species share the re-arranged organisation of the tRNA cluster. They feature the arrangement Gln (rev), Ile (fw), Met (fw) (), and display non-conserved spacers of 10–47 nucleotides between the tRNA genes (). All non-Benthophilinae species on the other hand feature the arrangement Ile (fw), Gln (rev), Met (fw) without spacers ().
Figure 1. Top: Origin of the re-arranged tRNA cluster Gln, Ile, Met. Most Gobiidae carry the arrangement Ile, Gln, Met without spacers. Benthophilinae however carry the arrangement Gln, Ile, Met, and feature variable length spacers between the genes. Bottom: Alignment of Benthophilinae tRNA sequences. Spacer sequences vary between species, but display more similarities among Neogobius species (Neogobius melanostomus, Neogobius fluviatilis) and Ponticola species (Proterorhinus semilunaris, Babka gymontrachelus, Ponticola kessleri), respectively.
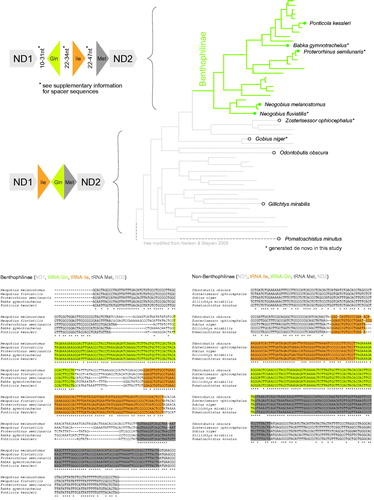
This change in gene arrangement yields further evidence for an evolutionary radiation at the root of the Benthophilinae (Neilson and Stepien Citation2009a). The spacer sequences display substantial divergence between species (). Since spacer sequences are more similar among the two Neogobius species and among the three Proterorhinus, Babka and Ponticola species,respectively, they support the previously observed division of Benthophilinae into a Neogobius and a Ponticola lineage (Neilson and Stepien Citation2009a; Medvedev et al. Citation2013). The spacer sequences appear to evolve quite freely and may be useful to (1) clarify currently debated relationships among Ponticola species (Neilson and Stepien Citation2009a; Medvedev et al. Citation2013) and to (2) confirm the identity of putative cryptic tubenose goby species (Neilson and Stepien Citation2009b).
Using long read technology, we investigated the origin of a recently detected 1250 bp sequence insertion in the round goby mitochondrial genome (Adrian-Kalchhauser et al. Citation2017), and found that individuals from across the native and invasive range carry the insert (). Accordingly, the insert did not arise recently during the invasion, and was not maintained by drift, but rather constitutes a genuine feature of the Neogobius melanostomus mitochondrial genome. This incites questions on its potential functional significance and potential reasons for retention in an otherwise economized mitochondrial genome. Expression analyses and replication assays may reveal whether the sequence plays a role in transcription or replication of the round goby mitochondrial genome.
Figure 2. Global occurrence of the novel sequence insertion. (A) Map of sampling sites in North America (St. Lawrence river) and Europe (Rhine river, Baltic Sea, and native region Black Sea). (B) Density of single nucleotide polymorphisms observed in the novel round goby non-coding insert, the adjacent D-loop, and adjacent expressed coding and non-coding sequences.
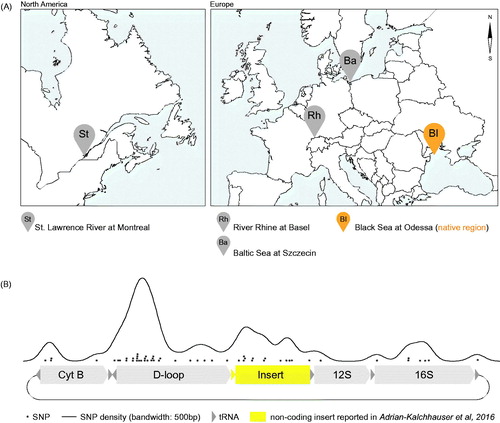
To understand whether the inserted sequence experiences mutation restraints, we quantified the number of single nucleotide polymorphisms (SNPs) detected between the four analysed mitochondrial genomes. We find fewer SNPs within expressed sequences (Cytochrome B, 12S and 16S) than in the D-loop and the sequence insertion (). Accordingly, a function of the insert that requires sequence conservation is unlikely. We find that SNPs accumulate in the D-loop following the TAS sites (Adrian-Kalchhauser et al. Citation2017), indicating the presence of a hypervariable region in the round goby control region similar to human Hypervariable Segments I and II (Stoneking Citation2000).
Hypervariable sites are potentially useful markers for studies focusing on short time evolutionary frames. Mutation rates in human hypervariable segments are four to six times higher than at average positions (Meyer et al. Citation1999). The round goby hypervariable regions may therefore be a useful source of haplotypes in future phylogenetic studies. Previously, the region has been challenging to sequence due to its highly repetitive nature. Our long-read approach circumvents this issue and highlights the power of single molecule long-read sequencing for short-term mitochondrial phylogenies and for biogeographical analyses.
Author contributions
IAK and SG designed research, IAK and SG organized samples, SG performed lab experiments, JCW, SG and IAK analysed data and wrote the manuscript.
Permissions
Fish used in this work were caught in accordance with permission 2-3-6-4-1 from the Cantonal Office for Environment and Energy, Basel Stadt.
Acknowledgements
We are grateful to Yuriy Kvach, Mariella Rasotto, Alexander Cerwenka, Przemysław Czerniejewski, and Pierre Magnan for providing tissue samples and to Silvia Kobel for technical support.
Disclosure statement
The authors have no conflict of interest to declare.
References
- Adrian-Kalchhauser I, Svensson O, Kutschera VE, Alm Rosenblad M, Pippel M, Winkler S, Schloissnig S, Blomberg A, Burkhardt-Holm P. 2017. The mitochondrial genome sequences of the round goby and the sand goby reveal patterns of recent evolution in gobiid fish. BMC Genomics. 18:177.
- De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. NanoPack: visualizing and processing long-read sequencing data . Bioinformatics. 34:2666–2669.
- Hirsch PE, N'Guyen A, Adrian-Kalchhauser I, Burkhardt-Holm P. 2015. What do we really know about the impacts of one of the 100 worst invaders in Europe? A reality check. Ambio. 45(3):267–279.
- Kornis MS, Mercado-Silva N, Vander Zanden MJ. 2012. Twenty years of invasion: a review of round goby Neogobius melanostomus biology, spread and ecological implications. J Fish Biol. 80:235–285.
- Medvedev DA, Sorokin PA, Vasil’ev VP, Chernova NV, Vasil’eva ED. 2013. Reconstruction of phylogenetic relations of Ponto-Caspian gobies (Gobiidae, Perciformes) based on mitochondrial genome variation and some problems of their taxonomy. J Ichthyol. 53:702–712.
- Meyer S, Weiss G, von Haeseler A. 1999. Pattern of nucleotide substitution and rate heterogeneity in the hypervariable regions I and II of human mtDNA. Genetics. 152:1103–1110.
- Neilson ME, Stepien CA. 2009a. Escape from the Ponto-Caspian: evolution and biogeography of an endemic goby species flock (Benthophilinae: Gobiidae: Teleostei). Mol Phylogenet Evol. 52:84–102.
- Neilson ME, Stepien CA. 2009b. Evolution and phylogeography of the tubenose goby genus Proterorhinus (Gobiidae: Teleostei): evidence for new cryptic species. Biol J Linnean Soc. 96:664–684.
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol. 29:24–26.
- Roche KF, Janac M, Jurajda P. 2013. A review of Gobiid expansion along the Danube-Rhine corridor - geopolitical change as a driver for invasion. Knowl Manag Aquat Ecosyst. 411:01.
- Stoneking M. 2000. Hypervariable sites in the mtDNA control region are mutational hotspots. Am J Human Genet. 67:1029–1032.
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration . Brief Bioinform. 14:178–192.