
Abstract
Magnolia odoratissima Law et R. Z. Zhou only produced in Yunnan Province of China. The species with extremely small populations and critically endangered, and now is affirmed as the second most important protection of wild plants in China. In this study, the complete chloroplast genome (cp) of M. odoratissima was sequenced, assembled and analyzed. Our results indicated that the cp genome of M. odoratissima was 159,536 bp in length, containing a large single copy (LSC, 87,688 bp) region and a small single copy (SSC, 18,764 bp) region separated by two reduced inverted repeats (IRs, 26,543 bp). A total of 114 unique genes were identified, including 80 protein-coding genes, 30 transfer RNA (tRNA) genes and 4 ribosomal RNA (rRNA) genes. The overall GC content of M. odoratissima cp genome was 39.3%. Most of the 88 simple sequence repeats (SSRs) were mononucleotides motifs of A/T types and were found to be located in non-coding regions. Phylogenetic analysis of M. odoratissima showed that M. odoratissima was the base group of genus Magnolia with strongly bootstrap supported.
Magnolia odoratissima Law et R. Z. Zhou is an ornamental shrub species, which belongs to the genus Magnolia of the Magnoliaceae. M. odoratissima sparsely distributed in limestone habitats of Xichou, Maguan and Malipo counties in Yunnan Province of China. In recent years, the nature habitat of M. odoratissima was severely damaged by over-exploitation, Field survey showed that M. odoratissima population fell sharply and now only five populations were remained on limestone mountains (He et al. Citation2018). It is the extremely small population species endemic to Yunnan province and ‘Endangered’ species in IUCN Red List (http://www.iucnredlist.org/). The age grade of M. odoratissima is relatively stable. The main reason for the decline of population is lack of illumination, the low seed yield and the competition among the associated tree species (He et al. Citation2018). It is urgent to take effective measures to protect this endangered and endemic species based on field investigation and scientific research. In addition, its phylogenetic position is still unclear in previous studies because of the lack of sampling. Here we characterized the complete chloroplast (cp) genome sequence of M. odoratissima based on the genome skimming sequencing data, detected the occurrence, type and distribution of simple sequence repeats (SSRs) and constructed the phylogenetic tree based on the maximum-likelihood (ML) method.
The fresh leaves of M. odoratissima were collected from the original habitat of Xichou district in Yunnan province (104°30′30″E, 23°12′50″N). The voucher specimen was deposited at Herbarium, Kunming Institute of Botany, CAS (KUN). Total genomic DNA was isolated from fresh leaves using the modified cetyltrimethyl ammonium bromide (CTAB) method (Doyle Citation1987) to construct chloroplast DNA libraries. The extracted DNA was sequenced using the Illumina Miseq platform (Illumina, San Diego, CA, USA). In all, 177 M of 150-bp raw paired reads were retrieved. Prior to chloroplast de novo assembly, low-quality reads were filtered out, and resultant clean reads were assembled using the CLC Genomic Workbench v10 (CLC bio, Aarhus, Demark). All the contigs were checked against the reference genome of Magnolia conifera (NC037001), using BLAST (https://blast.ncbi.nlm.nih.gov/) and aligned contigs were oriented according to the reference genome.
The genome was automatically annotated by using the CpGAVAS pipeline (Liu et al. Citation2012) and start/stop codons and intron/exon boundaries were adjusted in Geneious 8.1 (Kearse et al. Citation2012). The tRNA was identified through tRNAscan-SE v2.0 (Lowe and Chan Citation2016). Sequence data was deposited into GenBank. A physical map of the cp genome was generated using the online tool OGDraw v1.2 (http://ogdraw.mpimp-golm.mpg.de/) (Lohse et al. Citation2007). The complete cp genome sequence was submitted to the GenBank under Accession Number of MH795108.
The complete cp genome of M. odoratissima was 159,536 bp in length. It was the typical quadripartite structure, in which two inverted repeats (IRs, 26,543 bp) were separated by the large single-copy (LSC, 87,688 bp) and the small single-copy (SSC, 18,764 bp) regions. The cp genome had 114 genes, including 80 protein-coding genes (PCGs), 30 tRNA genes, and 4 rRNA genes. Most of genes occurred as single-copy, while PCGs (ndhB, rpl2, rpl23, rps12, rps7, ycf15, ycf2), seven tRNAs (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, trnV-GAC) and four rRNA genes (rrn16, rrn23, rrn4.5, rrn5) had two copies. In addition, two PCGs (clpP and ycf3) had two introns each; nine PCGs (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16 and rpoC1) contained one intron. The overall GC content of the cp genome was 39.3%, while that of LSC, SSC and IR regions was 38.0%, 34.3% and 43.2%, respectively. Phobos v3.3.12 (Leese et al. Citation2008) and SSRHunter (Li and Wan Citation2005) was used to find the SSR markers present in the chloroplast genome of M. odoratissima. It uses a recursive algorithm to search for repeats with lengths between one and six. The unit sizes of mono-, di-, tri-, tetra-, penta-, hexa-nucleotide repeats were set to minimum number of repeats of 10, 5, 4, 3, 3, 3, respectively. We removed one inverted repeat region (IRa) in SSRs analysis and manually checked the detected repeats. The occurrence and distribution of different types of SSR in the cp genome were summarized in . A total of 88 SSRs were detected throughout the cp genome of M. odoratissima, with 69, eight, two, six, one and two for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide repeats, respectively. The majority of the mono-nucleotides were A or T and all of di-nucleotides were AT or TA repeats, which was consistent with the A/T-richness in the complete cp genome (Xuan et al. Citation2013).
Table 1. Simple sequence repeats (SSRs) identified in the chloroplast genome of M. odoratissima.
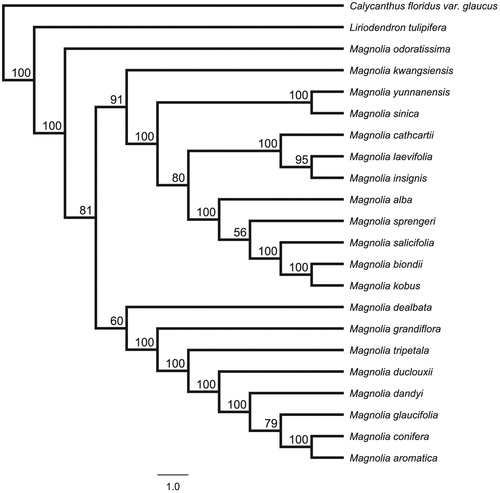
For maximum likelihood analyses, the complete chloroplast genome of 21 Magnolia species and one Liriodendron species were downloaded from NCBI GenBank. We chose Calycanthus floridus var. glaucus from Calycanthaceae as an outgroup. The combined datasets based on plastid genomes of 24 species were aligned by MAFFT v7.307 (Katoh and Standley Citation2013). A maximum-likelihood (ML) tree was constructed in RAxML (Stamatakis Citation2014) with the GTR + G model, and a total of 1000 bootstrap replicates were performed. The phylogenetic results showed that M. odoratissima was the base group of genus Magnolia with strongly bootstrap supported ().
Figure 1. Phylogenetic tree inferred by Maximum Likelihood (ML) method based on the complete chloroplast genome of 24 species, bootstrap values (%) are shown on the branch.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- He SL, Wang ZY, Wang W, Yang Y. 2018. Research on population of Magnolia odoratissima Law et R. Z. Zhou, a wild plant species with extremely small population. J West China For Sci. 47:64–68 (Chinese).
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647.
- Leese F, Mayer C, Held C. 2008. Isolation of microsatellites from unknown genomes using known genomes as enrichment templates. Limnol Oceanogr Methods. 6:412–426.
- Li Q, Wan JM. 2005. SSRHunter: development of a local searching software for SSR sites. Hereditas. 27:808.
- Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, Guan X. 2012. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13:715.
- Lohse M, Drechsel O, Bock R. 2007. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 52:267–274.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:W54–W57.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Xuan Y, Lei G, Bo W, Su YJ, Wang T. 2013. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of Cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 5:688–698.