
Abstract
Scurrula parasitica L., a hemiparasitic plant species, is a medicinal plant reported to have anticancer effect on human cancer cell lines. The whole chloroplast (cp) genome sequence of Scurrula parasitica has been characterized from Illumina pair-end sequencing. The complete cp genome is 121,750 bp in length, containing a large single copy (LSC) region of 70,270 bp and a small single copy (SSC) region of 6,106 bp, which are separated by a pair of 22,687 bp inverted repeat (IR) regions. The genome contains 106 genes, including 66 protein-coding genes, 28 tRNA genes, 8 ribosomal RNA genes and 4 pseudogenes. The overall GC content of S. parasitica cp genome is 37.2%, while the corresponding values of the LSC, SSC and IR regions are 34.4%, 25.9% and 42.9%, respectively. Phylogenetic analysis revealed that S. parasitica is closely related to Taxillus (3 spp.), with strong support values.
Scurrula parasitica L. is a medicinal plant reported to have anticancer effect on human cancer cell lines (Ali et al. Citation2013), and grown as hemiparasite on a wide range of hosts, including species of Apocynaceae, Euphorbiaceae, Fabaceae, Fagaceae, Lythraceae, Moraceae, Punicaceae, Rosaceae, Rutaceae, Sapindaceae, Theaceae and Ulmaceae. To facilitate its genetic research and contribute to its utilization, in this study, we assembled and characterized the complete chloroplast genome sequence of S. parasitica based on the Illumina pair-end sequencing data, which will be helpful for further studies on its chloroplast genetic engineering.
Fresh leaves of S. parasitica were collected on a host tree species, Ligustrum lucidum in the Wangjiang campus of Sichuan University (E104.087985°, N30.630763°; voucher specimen SZ-00545051 is deposited at the herbarium of Sichuan University (SZ) in Chengdu of Sichuan province, China), and immediately stored in liquid nitrogen below −80 °C. Total genomic DNA were extracted with a modified CTAB method (Doyle Citation1987). A total of 22 million 150-bp raw pair-end reads were yielded by an Illumina Hiseq 2500 platform (Illumina, San Diego, CA). The Trimmomatic (Bolger et al. Citation2014) was used to filter the raw reads and get high-quality clean reads. We used NOVOPlasty v2.6.3 (Dierckxsens et al. Citation2017), Geneious v8.1.4 and Velvet (Zerbino and Birney Citation2008) to assemble S. parasitica. We annotated the plastid genomes using Plann v1.1 (Huang and Cronk Citation2015) and corrected the annotation with Geneious v8.1.4 (Kearse et al. Citation2012) and Sequin v13.70 (http://www.ncbi.nlm.nih.gov/Sequin/). The complete cp genome sequence of S. parasitica was deposited in GenBank under accession number MH101514.
The S. parasitica cp genome is 121,750 bp in length, exhibits a typical quadripartite structural organization, consisting of a large single copy (LSC) region of 70,270 bp, 2 inverted repeat (IR) regions of 22,687 bp and a small single copy (SSC) region of 6,106 bp. The genome contains 106 genes, including 66 protein-coding genes, 28 tRNA genes, 8 ribosomal RNA genes and 4 pseudogenes. The most of gene species occur as a single copy, while 15 gene species occur in double copies. The overall GC content of S. parasitica cp genome is 37.2%, while the corresponding values of the LSC, SSC, and IR regions are 34.4%, 25.9%, and 42.9%, respectively.
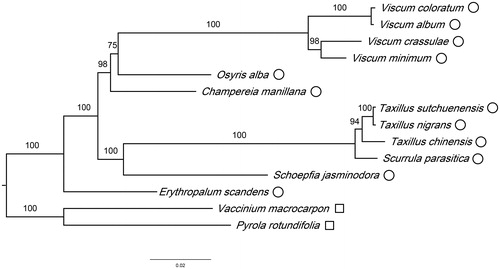
To infer the phylogenetic relationships of Santalales, we constructed a phylogenetic tree based on another 11 Santalales species’ cp genomes. Cp genomes data for those 11 species were downloaded from the NCBI database, including Champereia manillana (NC_034931.1), Erythropalum scandens (NC_036759.1), Osyris alba (NC_027960.1), Schoepfia jasminodora (NC_034228.1), Taxillus chinensis (NC_036306.1), T. sutchuenensis (NC_036307.1), T. nigrans (MH095982), Viscum album (NC_028012.1), V. coloratum (NC_035414.1), V. crassulae (NC_027959.1), and V. minimum (NC_027829.1). Two holoparasitic species of Ericales (Pylora rotundifolia KU833271.1 and V. macrocarpon NC_019616.1) were adopted as outgroups. The amino acid sequences of proteins encoded by their common genes were extracted and aligned using MAFFT V7.158 (Katoh and Standley Citation2013) and MEGA v6.0 (Tamura et al. Citation2013). We used the RAxML V8.2.11 software (Stamatakis Citation2014) to construct a ML tree. Bootstrap analysis was performed with 1000 replicates. Then we used FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/) to check the results.
Phylogenetic analysis demonstrated that 11 out of the 12 species of Santalales clustered into two highly-supported clades, Viscum (4 spp.) and Osyris (Santalaceae) plus Champereia (Opiliaceae) formed one clade, while Scurrula (Loranthaceae) and Taxillus (3 spp.) plus Schoepfia (Schoepfiaceae) formed another clade, whereas Erythropalum (Erythropalaceae) was the most distant clade within the Santalales (). Although the monophyly of Viscum was strongly supported, the monophyly of Santalaceae, as implied by the apparent sister relationship between Viscum (Visceae) and Osyris (Santaleae), received a moderate bootstrap support (75%) and merits closer investigation.
Figure 1. Phylogenetic relationships of Santalales species using whole chloroplast genome. GenBank accession numbers: Champereia manillana (NC_034931.1), Erythropalum scandens (NC_036759.1), Osyris alba (NC_027960.1), Schoepfia jasminodora (NC_034228.1), Taxillus chinensis (NC_036306.1), T. sutchuenensis (NC_036307.1), T. nigrans (MH095982), Viscum album (NC_028012.1), V. coloratum (NC_035414.1), V. crassulae (NC_027959.1) and V. minimum (NC_027829.1).
In summary, the complete cp genome of S. parasitica do not only provides important insight into its genome structure and composition, but also plays a critical role in constructing phylogeny of the Santalales.
Acknowledgements
The authors are grateful to the opened raw genome data from public database.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Ali MA, Chanu KHV, Devi LI. 2013. Scurrula parasitica L.: a medicinal plant with high antioxidant activity. Int J Pharm Pharm Sci. 5:34–37.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Huang DI, Cronk QC. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30:2725–2729.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829.