
Abstract
Nymphaea capensis (Nymphaeaceae) is one of the species in subgenus Brachyceras, which usually found in tropical and subtropical areas. Here, we presented a complete chloroplast genome of Nymphaea capensis which is 159,998 bp long and has four subregions: 90,134 bp of large single copy (LSC) and 19,504 bp of small single copy (SSC) regions are separated by 25,180 bp of inverted repeat (IR) regions including 130 genes (85 protein-coding genes, eight rRNAs, and 37 tRNAs). The overall GC content of the chloroplast genome is 39.2% and those in the LSC, SSC, and IR regions are 37.8%, 34.5%, and 43.4%, respectively. Phylogenetic tree constructed with the whole chloroplast genomes present phylogenetic position of subgenus Brachyceras with available Nymphaea chloroplast genomes.
Nymphaea capensis is one of the species in subgenus Brachyceras, which usually found in tropical and subtropical areas of America, Africa, Asia, and Australasia (Mauve Citation1967). It was introduced from Africa to America, Asia, and Australia (Mauve Citation1967; La‐ongsri et al. Citation2009). Flowers of N. capensis are day-blooming and usually present blue color which is the main reason why its common name is Cape blue waterlily. Till now, there is no research showing phylogenetic position of N. capensis based on molecular sequences (Borsch et al. Citation2007; Borsch et al. Citation2008; Dkhar et al. Citation2010, Citation2013) supported by the fact that the number of N. capensis available sequences in NCBI is only eight (as of 30 October 2018). It indicates that complete chloroplast genome of N. capensis will be useful to unravel its accurate phylogenetic position together with available chloroplast genomes of other Nymphaea species including Nymphaea lotus (https://www.tandfonline.com/doi/full/10.1080/23802359.2018.1547154).
Here, we sequenced complete chloroplast genome of N. capensis (Sample deposited in the InfoBoss Cyber Herbarium (IN); IBS-00008) as a second chloroplast genome of subgenus Brachyceras after Nymphaea ampla (NC_035680; Unpublished). Total DNA of N. carpensis was extracted from fresh leaves by using a DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using HiSeq2000 at Macrogen Inc., Korea, and de novo assembly was conducted by Velvet 1.2.10 (Zerbino and Birney Citation2008), gap filling was done by SOAPGapCloser 1.12 (Zhao et al. Citation2011), and all bases were confirmed by alignment results generated by BWA 0.7.17 (Li Citation2013) and SAM tools 1.9 (Li et al. Citation2009). Geneious R11 v11.0.5 (Biomatters Ltd., Auckland, New Zealand) was used for chloroplast genome annotation.
The chloroplast genome of N. capensis (Genbank accession is MK111409) is 159,311 bp and has four subregions: 90,134 bp of large single copy (LSC) and 19,504 bp of small single copy (SSC) regions are separated by 25,180 bp of inverted repeat (IR). It contains 130 genes (85 coding genes, 8 rRNAs, and 37 tRNAs); 17 genes (6 coding genes, 4 rRNAs, and 7 tRNAs) are duplicated in IR regions. The overall GC content of N. lotus is 39.2% and those in the LSC, SSC, and IR regions are 37.8%, 34.5%, and 43.4%, respectively.
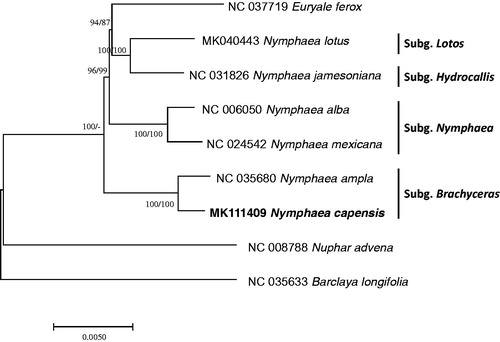
Nine complete Nymphaeaceae chloroplast genomes covering six Nymphaea species were used for constructing two phylogenetic trees; neighbor joining and maximum parsimony methods using MEGA X (Kumar et al. Citation2018). Multiple sequence alignment was conducted MAFFT 7.388 (Katoh and Standley Citation2013). The phylogenetic tree confirmed that two species, N. capensis and N. ampla, in subgenus Brachyceras were clustered into one clade (). However, Euryale ferox was nested in the clade of Nymphaea which is same to the previous study of N. lotus chloroplast genome (https://www.tandfonline.com/doi/full/10.1080/23802359.2018.1547154) and different from the previous study (Borsch et al. Citation2007). It shows that additional Nymphaea chloroplast genomes will make clear phylogenetic relationships of Nymphaea subgenera.
Figure 1. Neighbor joining and maximum parsimony phylogenetic tree (bootstrap repeat is 10,000) of nine Nymphaeaceae chloroplast genomes including one outgroup species: Nymphaea capensis (MK111409, this study), Nymphaea lotus (MK040443), Nymphaea ampla (NC_035680), Nymphaea jamesoniana (NC_031826), Nymphaea mexicana (NC_024542), Nymphaea alba (NC_006050), Euryale ferox (NC_037719), Barclaya longifolia (NC_035633), and Nuphar advena (NC_008878). The numbers above branches indicate bootstrap support values of neighbor joining and maximum likelihood tree, respectively.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Borsch T, Hilu KW, Wiersema JH, Löhne C, Barthlott W, Wilde V. 2007. Phylogeny of Nymphaea (Nymphaeaceae): evidence from substitutions and microstructural changes in the chloroplast trnT-trnF region. Int J Plant Sci. 168:639–671.
- Borsch T, Löhne C, Wiersema J. 2008. Phylogeny and evolutionary patterns in Nymphaeales: integrating genes, genomes and morphology. Taxon. 57:1052–1052.
- Dkhar J, Kumaria S, Rao SR, Tandon P. 2010. Molecular phylogenetics and taxonomic reassessment of four Indian representatives of the genus Nymphaea. Aquat Bot. 93:135–139.
- Dkhar J, Kumaria S, Rao SR, Tandon P. 2013. New insights into character evolution, hybridization and diversity of Indian Nymphaea (Nymphaeaceae): evidence from molecular and morphological data. Syst Biodivers. 11:77–86.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- La-ongsri W, Trisonthi C, Balslev H. 2009. A synopsis of Thai Nymphaeaceae. Nordic Journal of Botany. 27:97–114.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Preprint arXiv. 13033997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079.
- Mauve A. 1967. Water-lilies in South Africa. Fauna Flora (Transvaal). 18:31–35.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829.
- Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 12:S2.