
Abstract
Leigh syndrome (LS) is a mitochondrial progressive encephalopathy characterized by bilateral symmetric necrotic lesions of the central nervous system. Maternally inherited Leigh Syndrome (MILS) represents ∼10–20% of LS. Mutations in MT-ATP6 are the most common, being m.8993T > C/G the classical mutations. Molecular diagnosis for mitochondrial diseases is always a challenge and Multiplex ligation-dependent probe amplification (MLPA) of mitochondrial DNA can be an initial test for molecular diagnosis, although it is not widely used. We present a MILS patient in which MLPA was able to detect the common m.8993 T > G mutation and serve as a first approach for the definite molecular diagnosis.
Keywords:
Introduction
Mitochondrial disorders are a heterogeneous group of diseases caused by genetically determined mitochondrial dysfunction. The mitochondrial proteome (∼1500 proteins) (Meisinger et al. Citation2008) has a dual genetic origin (mitochondrial and nuclear DNA). Mitochondrial DNA (mtDNA) is a ∼16.5kb circular DNA inherited exclusively from maternal mitochondria that encodes for 13 proteins along with 22 tRNAs and 2 rRNAs necessary for their transcription and translation. A cell can have 100 to1000 mitochondria, each of which can bear 2–10 mtDNA copies, and copies can differ from each other, feature that is known as ‘heteroplasmy’. The rest of the mitochondrial proteins are encoded by nuclear DNA and travel to the mitochondria after being traduced in the cytoplasm. Thus, mitochondrial diseases can be inherited through a maternal mtDNA or a Mendelian pattern.
Leigh syndrome (LS) is a mitochondrial progressive encephalopathy characterized by bilateral symmetric necrotic lesions of the central nervous system involving the brainstem and/or basal ganglia structures. Usually, disease onset is early in the first year of life, progression is quick and ends with the child’s life before the age of 3 (Rahman et al. Citation1996). In cases of late onset, disease progression can be slower (Wesolowska et al. Citation2015). A broad variety of genetic alterations can cause this disease, including nuclear and mtDNA mutations. More than 75 different genes have been associated with LS (Lake et al. Citation2016) and most of these genes encode for mitochondrial complex proteins or proteins crucial for their correct functioning.
Maternally inherited Leigh Syndrome (MILS) represents ∼10–20% of LS. Mutations in complex 1 (MT-ND1) and complex V (MT-ATP6) are the most common causes, being MT-ATP6 the most important (∼10%). The classical devastating MILS is caused by high heteroplasmic (>90%) mutations in the MT-ATP6 gene, being position m.8993 the mutation hot spot for m.8993T > G or T > C. When heteroplasmy is lower (∼70%) these mutations can cause a later onset form of the disease called NARP syndrome (neuropathy + ataxia + retinitis pigmentosa) (Bülent et al. Citation2012).
Molecular diagnosis for mitochondrial diseases is always a challenge. Guidelines suggest looking first in mtDNA for common mutations and deletions (Chi Citation2015; Parikh et al. Citation2015). Next-generation sequencing has become the first line approach in many parts of the world. However, in developing countries this is not available as a first step option. Multiplex ligation-dependent probe amplification (MLPA) (Schouten et al. Citation2002) can be used as an initial study for molecular characterization of mitochondrial diseases since it has been shown to be sensible enough to detect mutations.
Materials and methods
Patient consent and clinical information
Informed consent was obtained from the patient’s mother in order to publish this case report. Clinical data were gathered from the patient’s clinical records.
DNA isolation
Blood was withdrawn from the patient and her mother and frozen at −20 °C for 24–48h, after which it was thawed. Total DNA was extracted with a Cetyl Trimethylammonium Bromide (CTAB)/chloroform-isoamylic based protocol. Leukocytes were isolated with 3–4 T10E10 washes and then suspended in CTAB solution (2 g/l CTAB Sigma Aldrich®, St Louis, MO, 100mM Tris/HCl, 20mM EDTA, 1.4M NaCl, and 2% 2-mercaptoethanol) and incubated at 60 °C for 1 hour. Then chloroform-isoamyl alcohol solution (24:1) was added and the samples were centrifuged. The aqueous phase was collected into a new tube and mixed with three volumes of ice-cold 100% ethanol. Precipitated DNA was dissolved in T10E0.1 buffer and stored at −20 °C for further use.
mtDNA MLPA
This technique was performed on the patient and her mother’s leukocyte DNA using the P125-B1 SALSA MLPA kit (MRC-Holland®, Netherlands). One nanogram of total DNA was used as a template, previously treated with 1 µl of 0.5 mg/ml RNase (Biodynamics®, Argentina). After probe hybridization and ligation, PCR was carried out and the amplification products were run on an ABI3130 DNA analyzer (Applied Biosystems®), Foster City, CA)® As control samples, DNA from a pool of leukocytes from 10 healthy individuals was used. Peak plots of patient, mother, and control samples were visualized and normalized using GeneMarker software v. 1.75 (SoftGenetics®, State College, PA) to establish a dosage ratio of mutated mtDNAs.
An altered ratio was considered according to standard cut off values for MLPA analyses (1.3 and 0.7).
Sanger sequencing
A fragment of the MT-ATP6 gene was amplified using the following primer set (5′-3′): Forward AATGCCCTAGCCCACTTCTT (m.8893–8912) and Reverse TCATTAGGAGGGCTGAGAGG (9272–9288). Direct sequencing of the PCR products was performed using an ABI PRISM 3500 Genetic Analyzer (Applied Biosystems®).
Results
Patient’s clinical presentation
Baby girl from a non-consanguineous healthy couple who was presented at 3 months of age with central hypotonia, milestone regression, encephalopathy, hyperlactacidemia, and seizures. She also had a hypertrophic cardiomyopathy that required treatment with beta blockers. Her brain MRI showed symmetric necrotic lesions in the basal ganglia and brainstem and the spectroscopy revealed an important lactic acid peak. Her biochemical metabolic work-up showed increased lactic, pyruvic, succinic, and fumaric acids in the urine. Plasma amino acids revealed high Alanine with normal Citrulline. Blood acylcarnitines were unremarkable. Family history revealed a maternal half-brother that had had similar clinical findings and died at 7 months of age with no diagnosis. The patient’s clinical record plus the family history made MILS the most probable diagnosis.
Patient and mother’s genetic findings
As a first step in the approach to molecular diagnosis, mtDNA MLPA was performed on the patient’s leukocyte DNA. The probe that hybridizes with mtDNA between positions m.8957 and m.9023 did not amplify at all in the patient’s sample (). This made us speculate that there was a mutation in that region that was blocking the probe’s hybridization, ligation, and amplification. The next step was to sequence that region. Sanger sequencing of the patient’s MT-ATP6 gene revealed the mutation m.8993T > G in homoplasmy (). The mother’s DNA showed the same mutation in heteroplasmy ().
Figure 1. Patient and mother’s molecular characterization. Left column: patient’s studies; right column: mother’s studies. The large arrows shows the genetic diagnostic work-flow that was used in this case. (A and C) Patient and mother’s mtDNA MLPA Ratio chart analyzed by Gene Marker v. 1.75. Boxes in between the horizontal lines correspond to ratios for regions which are present in a normal copy number. Boxes below or above the horizontal lines correspond to ratios for regions which are deleted or duplicated in the analyzed sample. Black triangles: represent ratios for sequence mutated regions (m.3243A > G, m.3460G > A, m.8344A > G, m.11778G > A, and m.14484T > C). Small arrows point to the m.8957–m.9023 probe, found with no amplification signal in the patient and with normal amplification signal in the mother. Amplification signal of probes for two mutation-specific regions is observable. These probes (14484T > C and 8344A > G) can give a low signal in wild-type mtDNA and should have a 10-fold augmentation to consider the presence of a mutation, which is not seen in these cases. In the patient’s assay, the smallest amplified region (first left ratio) seems to be duplicated, this fragment corresponds to a sequence located in the D-loop, where the origin of replication of the heavy mtDNA strand begins, making it a three-strand structure, which can explain this finding. (B and D) Patient and mother’s MT-ATP6 Sanger sequencing: the small arrows point to the m.8993T > G mutation found in homoplasmy in the patient and heteroplasmy in the mother.
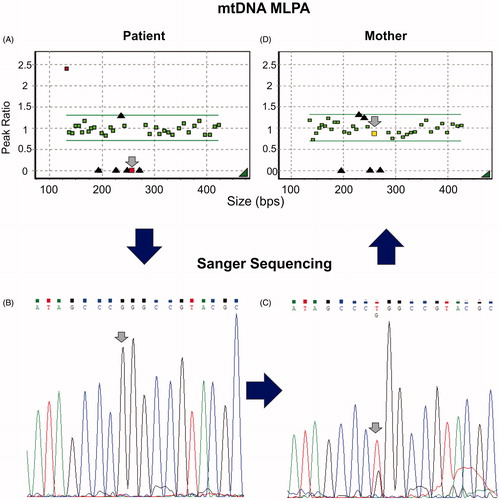
To see if we could determine the mother’s heteroplasmy level more accurately, we performed mtDNA MLPA on the mother’s DNA. The m.8957–m.9023 probe amplified in the same range as the controls’, concluding that the m.8993T > G mutation’s heteroplasmy level was undetectable with this technique (). Since mtDNA MLPA has been shown to detect a 20% heteroplasmy level (Mayorga et al. Citation2016), we assume that the mother’s heteroplasmy was below this value.
Discussion
MtDNA is still analyzed at a first approach for genetic diagnosis of mitochondrial diseases (Chi Citation2015). Even though the world is tending to use next-generation sequencing techniques from the onset, in developing countries this is still not possible due to the high cost and low availability. MLPA can be a good option to explore initially mtDNA (Kwon et al. Citation2011; Tońska et al. Citation2012), and here we see that it is sensible enough to detect a single base change. Even though the mtDNA MLPA commercial kit was not designed to detect the mutant m.8993 T > G/C DNA, it does detect the wild-type DNA of that region. Thus, low or lack of amplification of that MT-ATP6 probe should raise suspicion of a mutation in the region.
In conclusion, we propose mtDNA MLPA as a first line diagnostic tool for mitochondrial diseases as it is affordable, easy, and can detect mtDNA deletions and mutations in a single assay.
Acknowledgements
The authors thank the patient and her family for contributing to mitochondrial disease research.
Disclosure statement
The authors declare that they have no conflicts of interest.
Additional information
Funding
References
- Bülent K, Arıkan M, Maraş H, Abacı N, Çakıris A, Üstek D. 2012. Whole mitochondrial genome analysis of a family with NARP/MILS caused by m.8993T > C mutation in the MT-ATP6 gene. Mol Genet Metab. 107:389–393.
- Chi CS. 2015. Diagnostic Approach in Infants and Children with Mitochondrial Diseases. Pediatr Neonatol. 56:7–18.
- Kwon MJ, Ki CS, Kim JY, Lee ST, Kim JW, Kang SY. 2011. Multiplex ligation-dependent probe amplification (MLPA) assay for the detection of mitochondrial dna deletion in chronic progressive external ophthalmoplegia (CPEO). Ann Clin Lab Sci. 41:385–389.
- Lake NJ, Compton AG, Rahman S, Thorburn DR. 2016. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 79:190–203.
- Mayorga L, Laurito SR, Loos MA, Eiroa HD, de Pinho S, Lubieniecki F, Arroyo HA, Pereyra MF, Kauffman MA, Roqué M. 2016. Mitochondrial DNA deletions detected by multiplex ligation-dependent probe amplification. Mitochondrial DNA A DNA Mapp Seq Anal. 27:2864–2867.
- Meisinger C, Sickmann A, Pfanner N. 2008. The mitochondrial proteome: from inventory to function. Cell. 134:22–24.
- Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, Anselm I, Cohen BH, Falk MJ, Greene C, et al. 2015. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015. 17:689–701.
- Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. 1996. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann neurol. 39:343–351.
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. 2002. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30:e57.
- Tońska K, Piekutowska-Abramczuk D, Kaliszewska M, Kowalski P, Tańska A, Bartnik E, Pronicka E, Krajewska-Walasek M. 2012. Molecular investigations of mitochondrial deletions: evaluating the usefulness of different genetic tests. Gene. 506:161–165.
- Wesolowska M, Gorman GS, Alston CL, Pajak A, Pyle A, He L, Griffin H, Chinnery PF, Miller JA, Schaefer AM, et al. 2015. Adult onset Leigh syndrome in the intensive care setting: a novel presentation of a C12orf65 related mitochondrial disease. J Neuromuscul Dis. 2:409–419.