
Abstract
As a Korean endemic plant, Salix koriyanagi Kimura ex Goerz belongs to the genus Salix L. which is the largest genus of Salicaceae. In this study, we presented a complete chloroplast genome of S. koriyanagi which is 155,707 bp and has four sub-regions: 84,484 bp of large single copy (LSC) and 16,125 bp of small single copy (SSC) regions are separated by 27,549 bp of inverted repeat (IR) regions including 130 genes (83 coding regions, 8 rRNAs, and 38 tRNAs). The overall GC contents of the chloroplast genome were 36.7% and in the LSC, SSC, and IR regions were 34.4%, 31.00%, and 41.8%, respectively. A phylogenetic analysis showed that S. koriyanagi is distinct from Salix purpurea, which is incongruent to relationship based on their phenotypes.
Salix L. (Willow) is the largest genus of Salicaceae with about 450 species worldwide, occurring mainly in the Northern Hemisphere (Argus Citation1997). Salix koriyanagi Kimura ex Goerz is Korean endemic species usually located nearby streams (Lee Citation2003). According to Flora of China (Wu and Raven Citation1999), S. koriyanagi is similar to Salix purpurea L. based on several morphological characteristics: young branchlets, winter buds, and purplish red petioles. Based on the results of specimen observation, although S. purpurea is not distributed in Korean peninsula, morphology of S. koriyanagi and S. purpurea is almost same and they were clustered into one clade in the molecular phylogenic study (Wu and Raven Citation1999; Jin Citation2010).
Fresh leaves and specimen of S. koriyanagi were collected from Cheonggyecheon (Stream) in Seoul, Republic of Korea. Voucher specimens were deposited in InfoBoss Cyber Herbarium (IN); Y. Kim, IB-00566. Total DNA was extracted from fresh leaves of S. koriyanagi by using a DNeasy Plant Mini kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using HiSeq4000 at Macrogen Inc. in Republic of Korea. Raw reads were trimmed by Trimmomatic 0.33 (Bolger et al. Citation2014). We assembled chloroplast genome sequences with Velvet 1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser 1.12 (Zhao et al. Citation2011) to fill gaps, and confirmed all bases with conducting alignment against the assembled genome using BWA 0.7.17 and SAMtools 1.9 (Li et al. Citation2009; Li Citation2013). We used Geneious R11 11.0.5 (Biomatters Ltd., Auckland, New Zealand) to annotate S. koriyanagi chloroplast genome by transferring the annotations from the congeneric S. suchowensis sequence (NC_026772; Wu Citation2016). The S. koriyanagi chloroplast genome sequences were submitted to GenBank (MK120982).
The chloroplast genome of S. koriyanagi is 155,707 bp and has four sub-regions: 84,484 bp of large single copy (LSC) and 16,125 bp of small single copy (SSC) regions are separated by 27,549 bp of inverted repeat (IR). It contained 130 genes (83 coding regions, 8 rRNAs, and 38 tRNAs); 19 genes (eight coding regions, four rRNAs, and seven tRNAs) are duplicated in IR regions. The overall GC content of S. koriyanagi is 36.7% and in the LSC, SSC, and IR regions are 34.4%, 31.0%, and 41.8%, respectively.
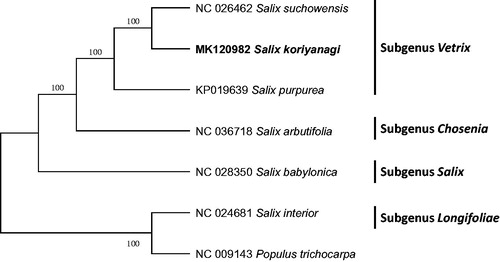
Sequence alignment of six Salix chloroplast complete genomes including S. purpurea (Wu Citation2015) was conducted by MAFFT 7.388 (Katoh and Standley Citation2013). The maximum parsimony (MP) tree was constructed with MEGA X (Kumar et al. Citation2018) with 1,000 bootstrap replicates. S. koriyanagi is positioned as a sister of a clade containing S. suchowensis and S. purpurea belonging to subgenus Vetrix (Lauron-Moreau et al. Citation2015) (). This chloroplast genome presents phylogenetic position between S. koriyanagi and S. purpurea with high bootstrap supports, of which phenotypes are similar to each other, suggesting S. koriyanagi is a clear endemic species in Republic of Korea.
Figure 1. Maximum parsimony phylogenetic tree (bootstrap repeat is 1,000) of six Salix chloroplast genomes and one outgroup species (Populus trichocarpa): Salix koriyanagi (MK120982), Salix suchowensis(NC_026462), Salix purpurea (KP019639), Salix arbutifolia (NC 036718), Salix babylonica (NC 028350), Salix interior (NC 024681), and Populus trichocarpa (NC 009143) as an outgroup species. The numbers above branches indicate bootstrap support values of maximum parsimony tree. Subgenera are presented in the right side of the tree with black bars.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
Related Research Data
References
- Argus GW. 1997. Infrageneric classification of Salix (Salicaceae) in the new world. Vol. 52, Syst Botany Monographs. 52:1–121.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120.
- Jin B. 2010. Analysis of the DNA barcode marker variation of Salix species in South Korea. Daegu (Korea): Daegu University.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 35:1547–1549.
- Lauron-Moreau A, Pitre FE, Argus GW, Labrecque M, Brouillet L. 2015. Phylogenetic relationships of American willows (Salix L., Salicaceae). PloS One. 10:e0121965
- Lee TB. 2003. Coloured flora of Korea.Hyang Mun Sa. Seoul. Korea.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079.
- Wu Z. 2015. The new completed genome of purple willow (Salix purpurea) and conserved chloroplast genome structure of Salicaceae. J Nat Sci. 1:e49.
- Wu Z. 2016. The whole chloroplast genome of shrub willows (Salix suchowensis). Mitochondrial DNA Part A. 27:2153–2154.
- Wu Z, Raven P. 1999. Flora of China. Vol. 4. Science.China.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829.
- Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 12:S2.