
Abstract
The Canada warbler (Cardellina canadensis) is a migratory neotropical warbler species of conservation concern. The 16,807 bp genome was sequenced and contains a 1224 bp control region, 37 genes, and has a nucleotide composition of 30.6% A, 23.7% T, 14.1% G, and 31.6% C. We constructed a phylogenetic tree of species in the order Passeriformes, which placed C. canadensis closest to species of Icteridae and Emberizidae. As the first complete mitogenome of any species in the family Parulidae, this mitogenome sequence provides a resource for future genetic and phylogenetic research involving C. canadensis and other Parulidae.
The Canada warbler (Cardellina canadensis) is a neotropical migrant that breeds in the northern forests of North America with fragmented trailing edge populations occurring at higher altitudes in the southern Appalachian mountains (Reitsma et al. Citation2009). In 2008, the Committee on the Status of Endangered Wildlife in Canada recognized the C. canadensis as threatened because of the species’ significant long-term decline (COSEWIC Citation2008). Habitat loss, specifically loss of older forest and forested riparian areas, has likely contributed to the decline of C. canadensis across a large portion of its range (Ball et al. Citation2016). While the conservation status and wide population distribution of C. canadensis make it an important study species, genetic studies have been limited. Additionally, to our knowledge, there are no complete mitochondrial genome sequences for any species in the Parulidae family. The complete mitogenome sequence will be useful for future research in conservation and population genetics as well as phylogenetics involving C. canadensis and other Parulidae.
We collected a blood sample and tissue biopsy from a C. canadensis specimen collected trapped in the Nantahala National Forest near Otto, NC in 2014 (35.050692 N, −83.512493 W). We extracted total genomic DNA using a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The remaining tissue biopsy is deposited in the Georgia Museum of Natural History accession number GMNHTC-13474. A library was constructed using a KAPA LTP Library Preparation Kit (KAPA Biosystems, Wilmington, MA) and sequenced with the Illumina® NextSeq system. We paired sequence reads by name and assembled a draft mitochondrial genome using Geneious version 8.1.2 (https://www.geneious.com, Kearse et al. Citation2012). We designed amplification and sequencing primers to re-sequence the complete mitochondrial genome by Sanger sequencing using an Applied Biosystems™ 3730xl DNA analyser (Applied Biosystems, Foster City, CA). Sanger reads were assembled into a complete mitochondrial genome, which we compared to the Illumina generated draft genome to review and resolve discrepancies. The mitogenome was annotated using Geneious version 11.0.5 (https://www.geneious.com, Kearse et al. Citation2012) and the complete sequence was deposited in GenBank (Accession number: MK033135). The complete mitochondrial genome of C. canadensis contains 16,807 nucleotides, one 1224 bp control region, 2 rRNA genes, 22 tRNA genes, and 13 protein-coding genes. The nucleotide composition of the genome is 30.6% A, 23.7% T, 14.1% G, and 31.6% C.
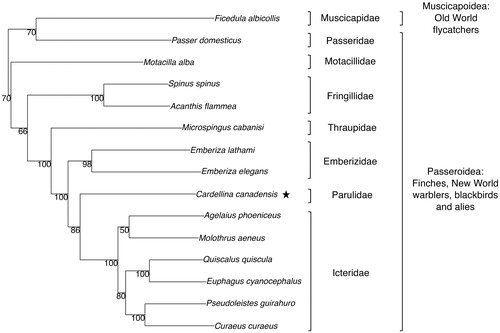
We constructed a phylogenetic tree of C. canadensis and other species in the order Passeriformes with a maximum likelihood analysis in R software (R Core Team Citation2016) using the package Phangorn version 2.4.0 (Schliep Citation2018) and following methods described in Schliep (Citation2011) (). We obtained complete mitochondrial genomes for our tree from GenBank. C. canadensis most closely clustered with six species from the family Icteridae and two from Emberizidae, and the clustering of species was consistent with taxonomical classifications. Our tree is consistent with the phylogenetic relationships of New World nine-primaried oscines observed by Bledsoe (Citation1988) and has a structure similar to more recent phylogenetic studies of this group (Klicka et al. Citation2000; Yuri and Mindell Citation2002).
Figure 1. Phylogenetic relationships among species in the Order Passeriformes inferred from complete mitochondrial genome sequences. The genome sequence accession numbers are as follows: Acanthis flammea (KR422696), Agelaius phoeniceus (JX516062), Cardellina canadensis (MK033135), Spinus spinus (HQ915866), Curaeus curaeus (JX516070), Emberiza elegans (KJ813903), Euphagus cyanocephalus (JX516072), Ficedula albicollis (NC_021621), Emberiza lathami (KX702277), Molothrus aeneus (JX516067), Motacilla alba (KT736087), Passer domesticus (CM004555), Microspingus cabanisi (KT272189), Pseudoleistes guirahuro (JX516071), Quiscalus quiscula (JX516064).
Acknowledgements
The authors gratefully acknowledge resources and funding provided by the University of Georgia Warnell School of Forestry and Natural Resources. We also acknowledge services provided by the Georgia Genomics and Bioinformatics Core. The specimen was collected under the University of Georgia Animal Use Permit A2017 02-019-Y2-A0.
Disclosure statement
The authors report no conflict of interest and are solely responsible for the content presented here.
Additional information
Funding
References
- Ball JR, Sólymos P, Schmiegelow FKA, Hache S, Schieck J, Bayne E. 2016. Regional habitat needs of a nationally listed species, Canada warbler (Cardellina canadensis), in Alberta, Canada. Avian Conserv Ecol. 11:10. Available from: http://www.ace-eco.org/vol11/iss2/art10/
- Bledsoe AH. 1988. Nuclear DNA evolution and phylogeny of the new world nine-primaried oscines. Auk. 105:504–515.
- COSEWIC. 2008. COSEWIC assessment and status report on the Canada Warbler Wilsonia Canadensis. Ottawa (ON): COSEWIC. www.sararegistry.gc.ca/status/status_e.cfm.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Klicka J, Johnson KP, Lanyon SM. 2000. New world nine-primaried oscine relationships: constructing a mitochondrial DNA framework. Auk. 117:321–336.
- R Core Team. 2016. R: a language and environment for statistical computing. https://www.r-project.org/.
- Reitsma L, Goodnow M, Hallworth M, Conway C. 2009. Canada warbler (Cardellina canadensis), version 2.0. In: Poole AF, editor. Birds of North America. Ithaca: Cornell Lab of Ornithology. doi:10.2173/bna.421.
- Schliep KP. 2011. Phangorn: phylogenetic analysis in R. Bioinformatics. 27:592–593.
- Schliep KP. 2018. Phangorn: phylogenetic reconstruction and analysis. R package version 2.4.0. https://cran.r-project.org/web/packages/phangorn/index.html.
- Yuri T, Mindell DP. 2002. Molecular phylogenetic analysis of Fringillidae, “new world nine-primaried oscines (aves: passeriformes).” Mol Phylogenet Evol. 23:229–243.