
Abstract
In this study, the complete mitochondrial genome of Graphium doson was recovered through Illumina sequencing data. This complete mitochondrial genome of G. doson is 15,285 bp in length and has a base composition of A (39.8%), T (41.1%), C (11.5%), and G (7.6%), demonstrating a bias of higher AT content (80.9%) than GC content (19.1%). The mitochondrial genome contains a typically conserved structure among Lineidae mitogenomes, encoding 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), two ribosomal RNA genes (12S rRNA and 16S rRNA), and a control region (D-loop region). The whole mt genome of G. doson and other Obtectomera mitogenomes (36 species, in total) were used for phylogenetic analysis. The result indicated G. doson has the closest relationship with Graphium leechi (KX011066.1) and clustered within Leptocircini Clade, which represents a distinct group.
The common jay, Graphium doson (Papilioninae: Leptocircini), is a black, tropical papilionid (swallowtail) butterfly with pale blue semi-transparent central wing bands that are formed by large spots. In this study, we provide a report of the complete mitochondrial genome of G. doson.
Larvaes of G. doson that collected from Lishui, Zhejiang Province, China (28°48'N, 119°91'E) were used in this experiment and maintained in lab environment. The DNA of 1 leg (dried or preserved at −20 °C) was extracted using a using QLAGEN DNEasy Extraction Kit following the manufactures instructions. The isolated DNA was stored in the sequencing company (HuiTong Tech, Shenzhen, China). Purified DNA was fragmented and used to construct the sequencing libraries following the instructions of NEBNext® Ultra™ II DNA Library Prep Kit (NEB, BJ, CN). Whole genomic sequencing was performed using the Illumina HiSeq 2500 Sequencing Platform (Illumina, CA, USA). Adapters and low-quality reads were removed using the NGS QC Toolkit (Patel and Jain Citation2012). Then assembly as implemented by SPAdes 3.11.0 (Bankevich et al. Citation2012). Circularization of this mt genome was confirmed using MITObim V1.9 (Hahn et al. Citation2013). The complete sequence was primarily annotated using ORF prediction in Unipro UGENE (Konstantin et al. Citation2012) combined with manual correction. All tRNAs were confirmed using the tRNAscan-SE search server (Lowe and Eddy Citation1997). Other protein-coding genes were verified using BLAST search on the NCBI website (http://blast.ncbi.nlm.nih.gov/), and manual correction for start and stop codons were conducted.
The complete mitochondrial genome of G. doson is 15,285 bp in length, which had been submitted to GenBank with accession No. MK144328. The entire genome consists of 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNA genes, is a typical mitogenome for butterfly. The overall base composition of the mitochondrial genome was 39.8% for A, 41.1% for T, 11.5% for G, and 7.6% for C, with a respectively higher AT content (80.9%). The length of 22 tRNAs that recognizing 20 amino acid range from 64 bp (trnM) to 78 bp (trnV).
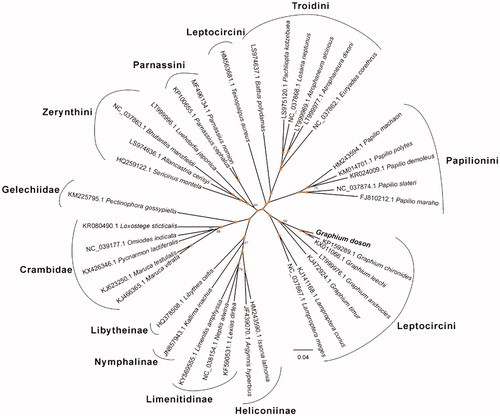
For phylogenetic analysis assessing the relationship of this mitogenome, we selected other 36 Obtectomera mitogenomes to construct genome-wide alignment. The genome-wide alignment of all mt genomes was done by HomBlocks (Guiqi et al. Citation2018) under the Gblocks trimming method, which only conserved phylogenetic informative positions in alignments, resulting in 4248 positions in total, including three conserved fragments shared by all mitogenomes. The whole genome alignment was analyzed using IQ-TREE version 1.6.6 (Lam-Tung et al. Citation2014) under the GTR + F + R5 model. The tree topology was verified under both 1000 bootstrap and 1000 replicates of SH-aLRT test. The resulting tree was represented and edited using FigTree v1.4.1. As shown in , the phylogenetic positions of these 37 mt genomes were successfully resolved with high bootstrap and SH-aLRT supports among almost all nodes. The result indicated G. doson has the closest relationship with Graphium leechi (KX011066.1) and clustered within Leptocircini Clade, which represents a distinct group.
Figure 1. Phylogenetic relationships among 37 mitochondrial genomes. The length of branch represents the divergence distance.
Disclosure statement
The authors declare that they have no competing interests. The authors alone are responsible for the content and writing of the paper.
References
- Bankevich A, Sergey N, Dmitry A, Gurevich AA, Mikhail D, Kulikov AS, Lesin VM, Nikolenko SI, Son P, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Guiqi B, Mao Y, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110:18–22.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucl Acids Res. 41:e129–e129.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 25:955–964.
- Lam-Tung N, Schmidt HA, von Haeseler A, Minh BQ. 2014. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Konstantin O, Golosova O. Mikhail Fursov and the UGENE team. 2012. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 28:1166–1167.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7:e30619