
Abstract
Vitis piasezkii is a widely distributed wild Vitis species in China with resistance to biological and non-biological stresses. In this study, the complete chloroplast genome of V. piasezkii was assembled for the first time. The chloroplast genome was 161,354 bp in length, containing a large single copy region (89,312 bp) and a small single copy region (19,309 bp) seperated by two inverted repeat regions. The genome harbors 135 genes, including 89 protein-coding genes, 38 tRNA genes, and eight rRNA genes. Phylogenetic tree analysis showed that V. piasezkii was the closest related to V. amurensis.
Vitis piasezkii is a widely distributed wild grape species in many provinces of China (Wan et al. Citation2008). With increasing human activities, the wild population of Vitis piasezkii is decreasing and some important resources of Vitis piasezkii may be destroyed. In the present study, we assembled the complete chloroplast genome of V. piasezkii for the first time (GeneBank: MK117077). This chloroplast genome will be benefical to elucidate V. piasezkii at the organelle level and to promote its conservation and utilization.
Fresh leaves were sampled from Research Vineyard for Grape Germplasm and Breeding in Shanghai Jiao Tong University, 800 Dongchuan Road, Minhang District, Shanghai, China (121°26′E; 31°02′N). DNA was extracted using the CTAB method (Qu et al. Citation1996) and stored in the Center for Viticulture and Enology, Shanghai Jiao Tong University. Sequencing the DNA by HiSeq X pair-end 150 sequencing platform (Illumina, CA, USA). In total, 3.65 Gb clean data were used to assemble the complete chloroplast genomic sequence using SOAPdenovo v2.04 (Luo et al. Citation2012). The chloroplast genome sequences of V. vinifira (DQ424856) were used as references. DOGMA package was used to predict genes (Wyman et al. Citation2004). BLAST 2.6.0+ (ftp.ncbi.nih.gov/blast/) was used to annotate genes.
Chloroplast length was 161,354 bp, included a large single copy region (1–89,312 bp,) and a small single copy region (115,674–134,983 bp), separated by inverted repeat region A and inverted repeat region B. The genome harbors 135 genes, including 89 protein-coding genes (PCGs), 38 tRNA genes, and eight rRNA genes. Among these genes, 16 genes have two copies, including eight PCGs (ndhB, rpl2, rpl23, rps19, rps7, ycf1, ycf2, and ycf15), four tRNAs (tRNA-Ala, tRNA-Asn, tRNA-Gly, and tRNA-Met), and four rRNAs (16S, 23S, 4.5S, and 5S), while four genes (tRNA-Arg, tRNA-Ser, tRNA-Thr, and tRNA-Val) have three copies, and two genes (tRNA-Ile and tRNA-Leu) have four copies.
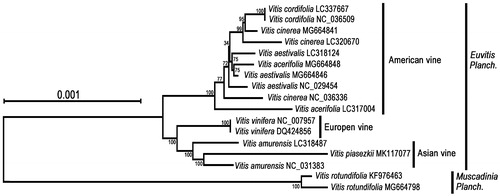
The chloroplast sequence of V. piasezkii was aligned with those of the 16 other Vitis species, using MAFFT version 7 (Katoh and Standley Citation2013). A neighbor-joining (NJ) phylogenetic tree was constructed by MEGA X (Kumar et al. Citation2018) (), which indicated that V. rotundifolia belongs to a clade different from the other Vitis species. Vitis species from Europe and Asia were more closely related than those from America. V. piasezkii was phylogenetically closer to V. amurensis. The phylogenetic relationships provide a basis for further study on the evolution of Vitis species in Asia.
Figure 1. The phylogenetic relationship of 17 species within the Vitis species based on neighbor-joining (NJ) analysis of chloroplast genomes (LSC, IRA, and SSC regions). The bootstrap values were based on 500 replicates and were shown next to the nodes.
Disclosure statement
There are no conflicts of interest for all the authors including the implementation of research experiments and writing this article.
Additional information
Funding
References
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 1:18.
- Qu X, Lu J, Lamikanra O. 1996. Genetic diversity in muscadine and American bunch grapes based on randomly amplified polymorphic DNA (RAPD) analysis. J Am Soc Hortic Sci. 121:1020–1023.
- Wan Y, Schwaninger H, Li D, Simon C, Wang Y, He P. 2008. The eco-geographic distribution of wild grape germplasm in China. Vitis. 47:77.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.