
Abstract
Malus sieversii (Rosaceae), a wild apple tree occurred in China’s Xinjiang province, is considered to be the ancestor of the modern cultivated apple. However, information on the chloroplast (cp) genome of this species is limited. With this study, we produced the first cp genome of M. sieversii using genome skimming. The whole cp genome was 163,230 bp long and comprised 128 genes, including 83 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The M. sieversii cp genome had a GC content of 35.6%. Phylogenetic analysis showed that M. sieversii was deeply nested within the Malus clade. This study will be useful for future studies on conservation genetics and potential applications in apple breeding.
Malus sieversii (Ledeb.) Roem, which is considered the ancestor of the modern cultivated apple, is a wild apple tree which mainly occurs in the Yili river valley in Xinjiang province, northwest China (Daccord et al. Citation2017; Duan et al. Citation2017). This species is of high economic value for apple breeding and is listed in the Chinese National Protected Plants list. However, the wild populations of M. sieversii experienced a dramatic decrease in the past years due to plant diseases and insect pests. Therefore, it is necessary to take effective measures in order to conserve this wild fruit tree species. Although numerous genetic studies investigated the species, M. sieversii, a comprehensive chloroplast (cp) genome is missing, which hampered our understanding of conservation genetics of this species at the cp genome level. In this study, we sequenced, assembled, and annotated the complete cp genome of M. sieversii, based on genome skimming.
Fresh leaves of M. sieversii were collected from Zhonghua Foushou Montain in Huocheng County (Xinjiang, China; 80.797527 E, 44.396994 N) and vouchers were deposited in the herbarium of Xinjiang University (accession number FSS 0015). DNA was extracted using the traditional CTAB method (Doyle and Doyle Citation1987). Whole genome sequencing was performed using an Illumina Hiseq platform in Suzhou, China and yielded 10.0 GB of raw data. After filtering, approximately 10 million clean high-quality reads were used for genome assembly. The complete circular genome was assembled using Velvet software (Zerbino and Birney Citation2008). After manual adjustment of the sequences, the genome was annotated using the Plastome Annotator (Huang and Cronk Citation2015). After this, a physical circular map of the genome with annotated genes was generated using the software OGDRAW (Lohse et al. Citation2013). A phylogenetic maximum likelihood (ML) tree was constructed using RaxML v. 8 software (Stamatakis Citation2014), based on an alignment of 14 Rosaceae cp genomes with Rosa multiflora and Fragaria x ananassa as outgroups (). The complete cp genome sequence of M. sieversii with detailed annotations was made available on GenBank under the accession number MH890570.
Figure 1. A phylogenetic tree based on 14 complete chloroplast genome sequences. Accession numbers: Rosa multiflora(NC039989); Malus hupehensis (MK020147); Malus prunifolia (NC031163); Malus micromalus(NC036368); Malus tschonoskii (NC035672); Malus florentina (NC035625); Malus trilobata (NC035671); Malus yunnanensis (NC039624); Malus baccata (KX499859); Prunus pedunculata (MG869261); Fragaria_x_ananassa_cultivar_Benihoppe (KY358226); Pentactina rupicola (NC016921); Crataegus kansuensis (NC039374); Malus sieversii (MH890570).
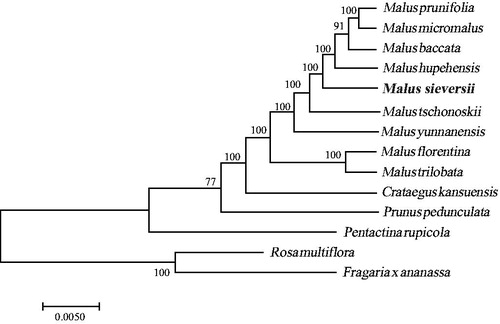
The cp genome of M. sieversii showed a typical quadripartite structure with a length of 163,230 bp and contained inverted repeats of 263,33 bp, separated by a large single copy and a small single copy of 88,396 bp and 19,196 bp, respectively. The cpDNA comprised 128 genes, with 83 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Twelve of the annotated genes contained one intron (rps12, trnL-UAA, atpF, trnK-UUU, rps16, rpl16, trnG-UCC, trnV-UAC, rpoC1, petB, petD, and ndhA), and six genes (ycf3, ndhB, trnA-UGC, trnI-GAU, rpl2, and clpP) contained two introns. The GC content of the cp genome was 35.6%. Furthermore, the phylogenetic analysis of 14 plastid genomes of Rosaceae showed that M. sieversii was deeply nested in the Malus clade (). The cp genome will be a valuable genetic resource and can be used for population genetic and phylogenetic studies of the genus Malus. Moreover, such information may be useful for future breeding of apple trees.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Daccord N, Celton J-M, Linsmith G, Becker C, Choisne N, Schijlen E, van de Geest H, Bianco L, Micheletti D, Velasco R, et al. 2017. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nature Genetics. 49:1099–1107.
- Duan NB, Bai Y, Song HH, Duan NB, Bai Y, Sun HH, Wang N, Ma YM, Li MJ, Wang X, Jiao C, Legall N, Mao LY, Wan SB, Wang K, He TM, Feng SQ, Zhang ZY, Mao ZQ, Shen X, Chen XL, Jiang YM, Wu SJ, Yin CM, Ge SF, Yang L, Jiang SH, Xu HF, Liu JX, Wang DY, Qu CZ, Wang YC, Zuo WF, Xiang L, Liu C, Zhang DY, Gao Y, Xu YM, Xu KN, Chao T, Fazio G, Shu HR, Zhong GY, Cheng LL, Fei ZJ, Chen XS. 2017. Genome re-sequencing reveals the history of apple and supports a two-stage model for fruit enlargement. Nature Communications. 8:249.
- Huang DI, Cronk QCB. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:w575–w581.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829.