
Abstract
Justicia leptostachya is an endemic species in China. The complete chloroplast genome sequence of J. leptostachya was generated by de novo assembly using whole genome next generation sequencing. The complete chloroplast genome was 150,898 bp in length, contained four parts: a large single copy (LSC) region of 82,585 bp, a small single copy (SSC) region of 17,477 bp and two inverted repeat (IRs) regions of 25,418 bp each. The genome annotation contained a total of 114 genes, including 80 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. The overall GC content was 38.5%. Phylogenetic analysis with the reported chloroplast genomes was also conducted.
Justicia leptostachya Hemsl. is a species of Acanthaceae mainly distributed in Guangdong, Guangxi and Hunan province in China (Hu et al. Citation2011). The whole plant of J. leptostachya can be used as traditional Chinese medicine for treating tuberous sclerosis without any side effects. Meanwhile, J. leptostachya is cultivated in many places because of its landscape value. In this research, the complete chloroplast genome of J. leptostachya was sequenced for the first time in order to provide more useful genome information to the chloroplast genome feature of Acanthaceae.
Total genomic DNA was extracted from leaves of J. leptostachya using a modified CTAB method (Li et al. Citation2013). The sample was collected from the greenhouse in South China Botanical Garden (113°21′6″E, 23°10′42″N, 30 m), Guangzhou, China. A voucher specimen was deposited at the South China Botanical Garden Herbarium. The Illumina paired-end sequencing library was constructed and high-quality pair-end (PE) reads were generated using the Illumina HiSeq 2500 platform. The PE reads were assembled using GetOrganelle pipeline (Jin et al. Citation2018). In order to get circular chloroplast DNA, the filtered plastid reads were transferred to the Bandage software (Wick et al. Citation2015) for visualization processing. After that, the complete plastid genome was annotated using Plastid Genome Annotator (PGA) (https://github.com/quxiaojian/PGA), then compared with the reference sequence of Andrographis paniculate artificially in Geneious 9.1.4 (Kearse et al. Citation2012). The final complete plastome was deposited into GenBank (accession number MK389502).
The circular complete cp genome of J. leptostachya is 150,898 bp in length, which consisted of one large single copy region of 82,585 bp, one small single copy region of 17,477 bp and a pair of inverted repeat regions of 25,418 bp. A total of 114 genes were predicted in the genome, including 80 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. The overall GC content was 38.5% and the GC contents of the LSC, SSC, and IR regions are 36.6%, 32.9%, and 43.5%, respectively.
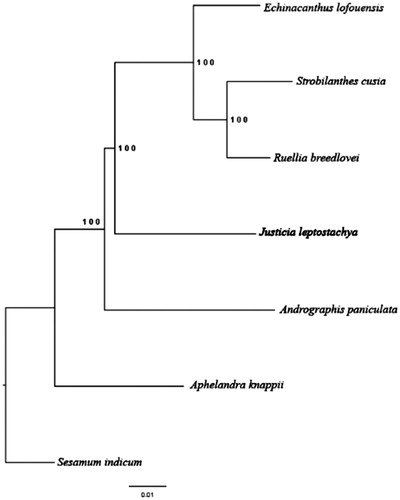
In order to understand the phylogenetic relationship between J. leptostachya and the related species of other genus in Acanthaceae, we generated a maximum-likelihood tree (ML) of seven species () by RAxML version 8.0.0 (Stamatakis Citation2014). The published complete genomes were downloaded from the NCBI GenBank database, including Andrographis paniculata (KF_150644), Echinacanthus lofouensis (MF_490441), Ruellia breedlovei (KP_300014), Strobilanthes cusia (MG_874806), Aphelandra knappiae (MH_909777), and Sesamum indicum (NC_016433) was hired as outgroup. Bootstrap (BS) value was calculated from 1000 replicate analysis. As expected, the results were consistent with the conclusion about Justicia by Kiel et al. (Kiel et al. Citation2017). This complete chloroplast genome can be subsequently used for population, phylogenetic, and cp genomic studies of the genus Justica.
Figure 1. ML phylogenetic tree of J. leptostachya with seven species was constructed using the chloroplast genome sequences. Numbers on the nodes are bootstrap values from 1000 replicates. Sesamum indicum was selected as outgroup.
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
Related Research Data
References
- Hu JQ, Deng YF, Daniel TF. 2011. Justicia. In: Wu ZY, Raven P, Hong DY, editors. Flora of China. Vol. 19. Beijing: Science Press; St. Louis: Missouri Botanic Garden Press; p. 449–461.
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv. 4:256479.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kiel CA, Daniel TF, Darbyshire I, McDade LA. 2017. Unraveling relationships in the morphologically diverse and taxonomically challenging “justicioid” lineage (Acanthaceae: Justicieae). Taxon. 66:645–674.
- Li JL, Wang S, Yu J, Ling W. 2013. A modified CTAB protocol for plant DNA extraction. Chin Bulletin Bot. 48:72–78.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31:3350–3352.