
Abstract
Chinese sorghum (Sorghum bicolor) is the most famous C4 plant and the most important cereal crops in the world. In this study, we obtained the complete chloroplast genome sequence of Chinese sorghum using BGISEQ-500 Sequencing Platform. Its chloroplast genome size has 141,266 bp, containing a large single copy region (83,406 bp), a small single copy region (13,712 bp), and a pair of IR regions (22,174 bp). The overall GC contents of the chloroplast genome were 38.5%. This circular genome contains 140 annotated genes, including 84 protein-coding genes, 48 tRNAs, and eight rRNAs. Further, the phylogenetic analysis using maximum likelihood (ML) method showed that Chinese sorghum has the closest phylogenetic relationship with Sorghum arundinaceum. This complete chloroplast genomes can be subsequently used for the genetic breeding and brewing industry.
Chinese sorghum (Sorghum bicolor) is the important of crop in the family Sorghum and one of the ancient cereal crops. Sorghum bicolor is a staple cereal in many parts of Africa, Asia, and America where it is grown mostly on small-scale, resource poor holdings (Too et al. Citation2018). Sorghum bicolor is grown for food, brewing, feed safety, and energy in both subsistence and commercial agriculture systems all over the world (Vermerris Citation2011). Among these cereal crop species, S. bicolor will replace corn and rice, resists high temperature, also has highest agronomic, nutritional, and economic value crop plant (Geoffrey et al. Citation2013). However, we know less of the origins of the chloroplast genome of Chinese sorghum C4 plant limited. In this study, we obtained the complete chloroplast genome of Chinese sorghum (S. bicolor) and explored the phylogenetic relationship with other species, which contributes to phylogenetic studies of these taxa and the brewing industry of this valuable species.
The specimen of S. bicolor was isolated from Yulin University test field in Yulin, Shanxi, China (109.73E; 38.29N). The total DNA of S. bicolor was extracted using Plant Genome DNA Extraction Kit (TIANGEN, Beijing, CA, CHN) and stored in Yulin University College of Life Science (No. YLUCLS01). The DNA sample was sequenced using the BGISEQ-500 Sequencing Platform (BIG, Shenzhen, CA, CHN). The chloroplast genome was assembled with SPAdes version 3.13 (Bankevich et al. Citation2012) and annotated using Blast and DOGMA (Wyman et al. Citation2004). The tRNA genes were further identified using tRNAscan-SE version 2.0 (Lowe and Chan Citation2016). The annotated chloroplast genome was submitted to GenBank database under accession No. NC543562.
The complete chloroplast genome of S. bicolor was a circle with 141,266 bp in size, containing a large single copy region (LSC) of 83,406 bp, a small single copy region (SSC) of 13,712 bp, and a pair of inverted repeat regions (IRs) of 22,174 bp. In total, 140 genes were annotated on this chloroplast genome, including 84 protein-coding genes (PCG), 48 transfer RNA genes (tRNA), and eight ribosomal RNA genes (rRNA). In the IR regions, a total of 18 genes were found duplicated, including five PCG species (rpl2, rpl23, rps7, rps12, and rps15), nine tRNA species (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnI-GAU, trnA-UGC, trnA-UGC, trnR-ACG, and trnN-GUU), and four rRNA species (16S rRNA, 23S rRNA, 4.5S rRNA, and 5S rRNA). The overall nucleotide composition is 30.9% A, 30.6% T, 19.2% C, and 19.3% G, with a total G + C content of 38.5%.
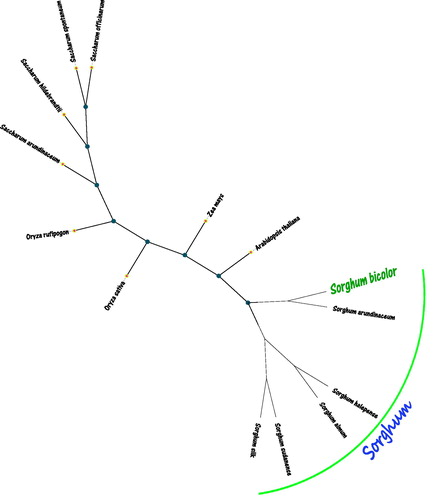
For phylogenetic relationships analysis, we selected other 13 plants chloroplast genomes from GenBank to assess the relationship of S. bicolor. The genome-wide alignment of all chloroplast genomes was constructed using HomBlocks (Bi et al. Citation2018). The phylogenetic trees were reconstructed using maximum likelihood (ML) methods. ML analysis were performed using RAxML version 8.2.4 (Stamatakis Citation2014), of which the bootstrap values were calculated using 5000 replicates to assess node support, and all the nodes were inferred with strong support using the ML methods. The final tree was represented and edited using MEGA version X (Kumar, Stecher et al. Citation2018). As shown in the phylogenetic tree (), the chloroplast genome of S. bicolor was the closest and clustered with Sorghum arundinaceum (No. LS398103.1).
Figure 1. The maximum likelihood (ML) tree inferred from Sorghum bicolor and other 13 plants chloroplast genomes. This tree was drawn without setting out groups. All nodes exhibit above 90% bootstraps. The length of branch represents the divergence distance. The other 13 plants species and corresponding GenBank accession numbers are as follows: Sorghum arundinaceum (LS398103.1), Sorghum halepense (LS398105.1), Sorghum almum (FJ650403.1), Sorghum sudanense (KC428145.1), Sorghum silk (FJ650401.1), Saccharum officinarum (NC035224.1), Saccharum arundinaceum (LC160130.1), Saccharum spontaneum (NC034802.1), Saccharum hildebrandtii (MF563371.1), Zea mays (NC001666.2), Oryza sativa (NC031333.1), Arabidopsis thaliana (NC000932.1), and Oryza rufipogon (KF359902.1).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110 18–22.
- Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD, Riera-Lizarazu O, Acharya CB, Mitchell SE, Harriman J, et al. 2013. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci. 110:453–458.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:54–57.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Too EJ, Onkware AO, Were BA, Gudu S, Carlsson A, Geleta M. 2018. Molecular markers associated with aluminium tolerance in sorghum bicolor. Hereditas. 155:20.
- Vermerris W. 2011. Survey of genomics approaches to improve bioenergy traits in maize, sorghum and sugarcane. J Integr Plant Biol. 53:105–119.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.