
Abstract
Eriobotrya japonica is the most important economic fruit crop with high edible, medical as well as ornamental values in China. In this study, we presented the complete chloroplast genome of E. japonica. The chloroplast genome size is 159,107 bp, containing a large single-copy region (87,213 bp), a small single-copy region (19,268 bp), and a pair of IR regions (26,313 bp). The overall GC contents of the chloroplast genome are 36.7%. Eriobotrya japonica whole chloroplast genome contains 129 genes, including 84 protein-coding genes, 37 tRNAs, and 8 rRNAs. Phylogenetic maximum likelihood (ML) analysis based on 15 plant species chloroplast genomes indicates that E. japonica is closely related to Eriobotrya bengalensis. These complete chloroplast genomes can be subsequently used for valuable species research of Eriobotrya.
Eriobotrya japonica is one of the most important subtropical characteristic fruits in China, which belongs to the Rosaceae family and origins in China and later was introduced to Mediterranean basin, Japan, Vietnam, and America (Blasco et al. Citation2014). It is also an important economic fruit crop in summer and has the high edible, medical as well as ornamental values. Eriobotrya japonica fruits have greatly appreciated by the people due to the good taste and leaves have been found naturally active compounds to treatment of diseases (Seong et al. Citation2018). However, our understanding of the chloroplast genome of the Eriobotrya plant is limited. In order to further study, the Eriobotrya and Rosaceae family genetic diversity and genetic structure of natural populations, we assembled and annotated the chloroplast genome that can provide much species the conservation and evolutionary studies. Here, we assembled the complete chloroplast genome of E. japonica and explored the phylogenetic relationship with other plant species, which contributes to phylogenetic studies and the collection and protection of genetic resources.
The specimen sample of E. japonica was isolated from Putian University test field in Putian (Fujian, China, 119.02E; 25.45N) and sample was deposited the Putian University. The total genomic DNA of E. japonica was extracted using the optimized CTAB method and stored in Putian University (No. PTU001). The total genomic DNA was used for the shotgun library construction and sequenced using the Illumina HiSeq 4000 Sequencing Platform System (Illumina Co., San Diego, CA). Quality control was performed to remove low-quality reads and adapters using the FastQC software (Andrews Citation2015). The chloroplast genome was assembled using the MITObim software (Hahn et al. Citation2013) and annotated using the OGDRAW online software (Lohse et al. Citation2013) and DOGMA software (Wyman et al. Citation2004). The tRNA genes were further identified using tRNAscan-SE software (Lowe Todd and Chan Citation2016). The annotated chloroplast genome sequence has been deposited into GenBank under the accession No. MH7888731.
The whole complete chloroplast genome of E. japonica was a circle with 159,107 bp in size, containing a large single-copy region (LSC) of 87,213 bp, a small single- copy region (SSC) of 19,268 bp, and a pair of inverted repeat regions (IRA and IRB) of 26,313 bp. The cpDNA of E. japonica comprised 129 genes, including 84 protein-coding genes (PCG), 37 transfer RNA genes (tRNA), and 8 ribosomal RNA genes (rRNA). In the IR regions, a total of 17 genes were found duplicated, including 6 PCG species (ycf2, ndhB, rpl2, rpl23, rps7, and rps12), 7 tRNA species (trnI-CAU, trnL-CAA, trnV-GAU, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU), and 4 rRNA species (rrn16, rrn23, rrn4.5, and rrn5). The overall nucleotide composition is: 31.2% A, 32.1% T, 18.7% C, and 18.0% G, with the total GC content is 36.7%.
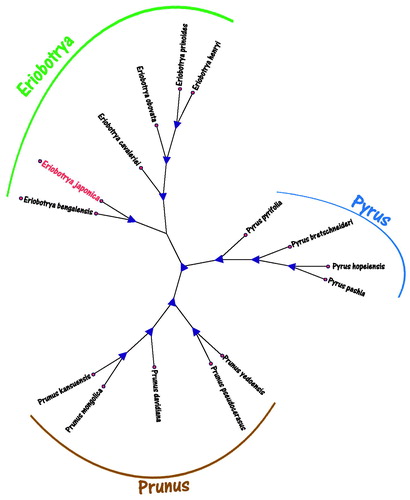
Phylogenetic maximum likelihood (ML) analysis, we selected other 14 plants species chloroplast genomes from GenBank to assess the relationship of E. japonica. The phylogenetic tree was reconstructed using ML methods. ML analysis was performed using the RaxML software (Stamatakis Citation2014), of which the bootstrap values were calculated using 2000 replicates to assess node support and all the nodes were inferred with strong support by the ML methods. The final tree was represented using the MEGA X software (Kumar et al. Citation2018) and edited using the iTOL online software(Letunic and Bork Citation2016). As shown in the phylogenetic ML tree result (), the chloroplast genome of E. japonica is clustered and closest with E. bengalensis and also closer to other Eriobotrya species in the evolutionary relationship. Simultaneously, the complete chloroplast of E. japonica is significance for the conservation and evolutionary studies.
Figure 1. The maximum likelihood (ML) phylogenetic tree of the 15 species from Eriobotrya japonica was constructed based on complete plants chloroplast genomes data. The analyzed species and corresponding GenBank accession numbers are as follows: Eriobotrya bengalensis (KJ170754.1), Eriobotrya henryi (KJ170752.1), Eriobotrya cavaleriei (KJ170759.1), Eriobotrya prinoides (KJ170757.1), Eriobotrya obovata (KJ170756.1), Pyrus pyrifolia (AP012207.1), Pyrus hopeiensis (MF521826.1), Pyrus pashia (NC034909.1), Pyrus bretschneideri (KX450881.1), Prunus kansuensis (NC023956.1), Prunus mongolica (NC037849.1), Prunus davidiana (NC039735.1), Prunus pseudocerasus (NC030599.1), and Prunus yedoensis (KU985054.1).
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
Related Research Data
References
- Andrews S. 2015. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Blasco M, Naval M. d M, Zuriaga E, Badenes ML. 2014. Genetic variation and diversity among loquat accessions. Tree Genet Genome. 10:1387–1398.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129–e129.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44:W242–W245.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Lowe Todd M, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:54–57.
- Seong NW, Seo HS, Kim JH, Kim YJ, Kim E, Lee JY, Ko JW, Kim JC. 2018. A 13-week subchronic toxicity study of an Eriobotrya japonica leaf extract in rats. J Ethnopharmacol. 226:1–10.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.