
Abstract
The complete mitochondrial genome of Common Greenshank Tringa nebularia was obtained using next-generation sequencing. The circular genome was 16,682 bp in length, consisting of 13 protein-coding genes, 22 transfer RNA genes, two ribosomal RNA genes, and a control region. The overall nucleotide composition was A: 31.8%, T: 24.9%, C: 29.8%, and G: 13.4%. Nine genes were encoded on the light strand, and the remaining 28 genes were encoded on the heavy strand. Most of the PCGs began with the ATG as the start codon, and four kinds of termination codons were used in this mitogenome. This study improves our understanding of the mitogenomic characteristics and its phylogenetic relationships within Charadriiformes.
The Common Greenshank Tringa nebularia (Charadriiformes, Scolopacidae) is full migratory with an extremely large distribution range, which has long greenish legs and a long bill with grey base, and show a white wedge on the back in flight. Recent studies of T. nebularia pay more attention on habitat ecology, geographical pattern, and breeding ecology (Thompson et al. Citation1986; Remisiewicz et al. Citation2014). However, the basic genetics data of T. nebularia is unclear. In this study, we sequenced the complete mitogenome of T. nebularia to better understanding the mitogenomic characteristics and its phylogenetic relationships within Charadriiformes.
The muscle specimen of T. nebularia was collected from the Lukou International Airport, Nanjing, Jiangsu Province, China (31°43′47″ N, 118°52′26″ E). The voucher specimen was preserved in absolute ethanol, which was stored at −20 °C in our laboratory at Jiangsu Open University, Jiangsu, China. Total DNA was extracted with standard phenol-chloroform methods (Sambrook et al. Citation1989). The sequencing libraries with average insert sizes of approximately 300 bp were prepared and then sequenced as 150 bp paired-end runs (about 8 Gb raw data) on the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA). The software of Geneious 9.1.4 was used for sequence quality analysis, data trimming, and de novo assembling (Kearse et al. Citation2012). The locations of the protein-coding genes, ribosomal RNAs (rRNAs), and transfer RNAs (tRNAs) were predicted using MITOS Web Server and identified by alignment with other mitogenome of Charadriiformes species (Bernt et al. Citation2013).
The circular mitogenome is 16,682 bp in length with 13 protein-coding genes (PCGs), two ribosomal RNAs (12S rRNA and 16S rRNA), 22 transfer RNA genes, and a non-coding region. The annotated mitogenome of T. nebularia has been deposited in GenBank (MK460251). The overall nucleotide composition was A: 31.8%, T: 24.9%, C: 29.8%, and G: 13.4%. Among the 37 genes, nine genes (tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, ND6, tRNAPro, and tRNAGlu) were encoded on the light strand, and the remaining 28 genes were encoded on the heavy strand. The total length of 13 protein-coding genes is 11,396 bp accounting for 68.31% of the complete genome. Most of the PCGs began with the ATG as the start codon.
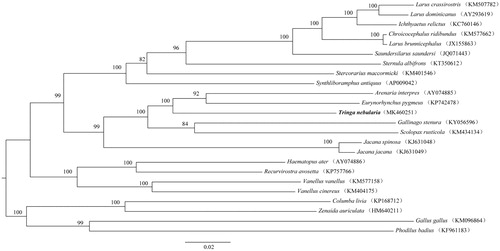
We performed a phylogenetic analysis based on 13 protein-coding genes (PCGs) of the available mitogenome sequences of Charadriiformes and four other species, using maximum likelihood (ML) algorithm in MEGA X (Kumar et al. Citation2018). The mitogenomes sequences were aligned using Muscle in MEGA X, and subsequently edited and trimmed (Kumar et al. Citation2018). Total of 11,322 bp were used for phylogenetic analyses. The phylogenetic analysis () resolved three well-supported clades of Charadriiformes, indicating great mitochondrial divergence within the Charadriiformes. This study improves our understanding of the evolution of mitochondrial DNA in Charadriiformes.
Figure 1. Phylogeny of T. nebularia and closely related 20 mitochondrial sequences constructed using the maximum likelihood (ML) method by analyzing 13 protein-coding genes (PCGs). Numbers above each branches are the ML bootstrap support. GenBank accession numbers of each species are shown in parentheses.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Remisiewicz M, Tree AJ, Underhill LG, Nowakowski JK. 2014. Geographical patterns in primary moult and body mass of Greenshank Tringa nebularia in Southern Africa. Ardea. 102:31–46.
- Sambrook J, Maniatis TE, Fritsch EF. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor (NY): Cold Spring Harbour Laboratory Press.
- Thompson DBA, Thompson PS, Nethersole-Thompson D. 1986. Timing of breeding and breeding performance in a population of greenshanks (Tringa nebularia). J Anim Ecol. 55:181–199.