
Abstract
In Korea, a new Bombyx mori (Lepidoptera: Bombycidae) strain, which has a peculiar larval body marking was bred to draw public attention to the importance of national resources. We sequenced the complete mitochondrial genome (mitogenome) of the strain “Hukpyobeom” and phylogenetic relationships of the strain were analyzed. The complete genome of the strain comprised 15,671 bp, contained a typical set of genes of animals, and had a gene arrangement and composition typical in Lepidoptera. The newly bred “Hukpyobeom” strain had a 20-50 bp longer whole genome size, 39 bp longer protein-coding genes, and 5-23 bp shorter intergenic spacer regions compared to the other strains. Phylogenetic analyses showed the “Hukpyobeom” to form a strong subgroup together with each one Japan- and China-originated strain. A strain originating from Russia (strain name Soviet_Union_No.1) was placed as the most basal lineage, forming a sister to the remaining strains, indicating genetic divergence within B. mori.
The domestic silkworm, Bombyx mori (Lepidoptera: Bombycidae), is an economically important resource; hence, more than 3000 different silkworm strains are maintained in Europe and Asia and these differ in diverse characteristics (Nagaraju Citation2000; Ryu et al. Citation2003). In Korea, approximately 300 silkworm strains are cultured annually and have been registered in the national genetic resource database. Recently, silkworm strains with peculiar outward appearances have become popular for educational and exhibition purposes and one such strain was bred several years ago in Korea (Kang et al. Citation2010; ). Therefore, obtaining a minimal but significant amount of genetic information for the strain is essential as a national genetic resource. Consequently, we sequenced the complete mitochondrial genome (mitogenome) of the strain, which would most likely provide enough genetic information to compare with those of the preexisting worldwide strains. This is particularly true in that previous study using DNA barcode revealed nearly identical sequences among the strains preserved in Korea (Kim et al. Citation2000).
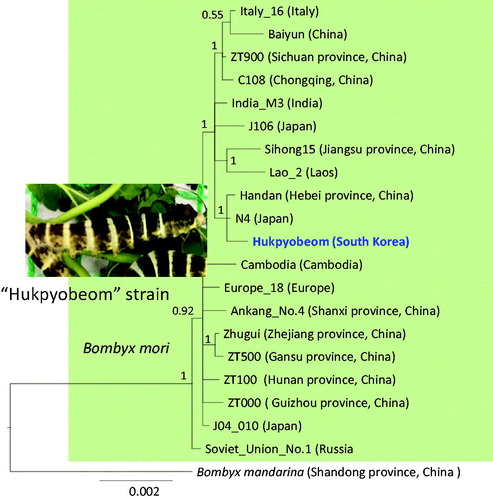
Figure 1. Phylogenetic tree of Bombyx mori. Bayesian inference (BI) method was used for phylogenetic analysis based on concatenated 13 protein-coding genes and 2 rRNA genes. The numbers at the node specify Bayesian posterior probabilities (BPP) by the BI method. The scale bar indicates the number of substitutions per site. The wild silkworm (Bombyx mandarina, FJ384796, Hu et al. Citation2010) was used as outgroup. GenBank accession numbers are as follows: Europe_18, GU966607 (Li et al. Citation2010); ZT500, GU966611 (Li et al. Citation2010); Zhugui, GU966609 (Li et al. Citation2010); ZT000, GU966613 (Li et al. Citation2010); J04_010, GU966612 (Li et al. Citation2010); Baiyun, KM279431 (Zhang et al. Citation2016); Italy_16, GU966596 (Li et al. Citation2010); C108, GU966630 (Li et al. Citation2010); ZT900, GU966600 (Li et al. Citation2010); India_M3, GU966595 (Li et al. Citation2010); Lao_2, GU966610 (Li et al. Citation2010); Sihong15, GU966617 (Li et al. Citation2010); Handan, GU966628 (Li et al. Citation2010); J106, GU966615 (Li et al. Citation2010); Ankang_No.4, GU966614 (Li et al. Citation2010); Cambodia, GU966601 (Li et al. Citation2010); ZT100, GU966603 (Li et al. Citation2010); and Soviet_Union_No.1, GU966599 (Li et al. Citation2010).
One larva of the “Hukpyobeom” strain (Jam 314) was obtained from the National Academy of Agricultural Science in Korea, and subsequently deposited at the Chonnam National University, Korea, under accession no. CNU8283. To sequence the mitogenome of “Hukpyobeom,” whole genome sequencing was performed using NextSeq-500 platform (Illumina, San Diego, CA, USA). Construction of the genome was conducted by de novo assembly. Owing to precise final genome sequence, no additional Sanger-based sequencing was conducted. Genomic sequences were compared with 18 silkworm strains reported in Li et al. (Citation2010) and one that was reported in Zhang et al. (Citation2016) and these originated from eight countries (China, Japan, India, Laos, Europe, Italy, Cambodia, and Russia). For phylogenetic analysis, 13 protein-coding genes (PCGs) and 2 rRNA genes were aligned, along with the outgroup species, B. mandarina. The Bayesian inference (BI) method was conducted using RAxML-HPC2 on XSEDE ver. 8.0.24 (Stamatakis Citation2014), implemented on the CIPRES Portal ver. 3.1 (Miller et al. Citation2010).
The complete 15,676 bp mitogenome of “Hukpyobeom” is composed of typical gene sets (2 rRNAs, 22 tRNAs, and 13 PCGs) and a major non-coding A + T-rich region (GenBank acc. no. MK613835). Among 13 PCGs only COI had a CGA codon that is frequently found in Lepidoptera (Park et al. Citation2016). The gene arrangement was identical to that of the ditrysian Lepidoptera that have the order trnM-trnI-trnQ between the A + T-rich region and ND2 (Kim et al. Citation2000). Compared to the 19 other country-preserving strains, “Hukpyobeom” had a 20–50 bp longer whole genome, 39 bp longer PCGs, and 5–23 bp shorter intergenic spacer region (Li et al. Citation2010; Zhang et al. Citation2016). A major difference in size stemmed from the number of microsatellite-like TA repeats located in the intergenic spacer region at ND3 and trnA junction and trnH and ND4 junction. In contrast, the length of tRNAs, rRNAs, and the A + T-rich region was highly similar.
Phylogenetic analyses showed that one strain originating from Russia (strain name Soviet_Union_No.1) was placed as the most basal lineage, forming the sister to the remaining strains, indicating a substantial genetic divergence within B. mori strains. The “Hukpyobeom” formed a strong subgroup together with each one Japan (strain name N4) and China-originated strain (Handan) with the highest nodal support. Also, other silkworm strains formed several subgroups with the highest nodal supports (). Probably such subgroups may have originated because of continuous inbreeding over a long period of time. We hope this mitogenome sequence of “Hukpyobeom” is useful for subsequent development of strain-diagnostic markers.
Disclosure statement
No potential conflicts of interest are reported by the authors.
Additional information
Funding
References
- Hu XL, Cao GL, Xue RY, Zheng XJ, Zhang X, Duan HR, Gong CL. 2010. The complete mitogenome and phylogenetic analysis of Bombyx mandarina strain Qingzhou. Mol Biol Rep. 37:2599–2608.
- Kang P-D, Jung I-Y, Kim K-Y, Kim M-J, Sohn B-H, Lee G-G. 2010. Breeding of two new silkworm varieties with peculiar laval mark, “Eolrukmal” and “Hukpyobeom”. Int J Indust Entomol. 20:115–116.
- Kim I, Bae JS, Sohn HD, Kang PD, Ryu KS, Sohn BH, Jeong WB, Jin BR. 2000. Genetic homogeneity in the domestic silkworm, Bombyx mori, and phylogenetic relationship between B. mori and the wild silkworm moth, B. mandarina, using mitochondrial COI gene sequences. Int J Indust Entomol. 1:9–17.
- Li D, Guo Y, Shao H, Tellier LC, Wang J, Xiang Z, Xia Q. 2010. Genetic diversity, molecular phylogeny and selection evidence of the silkworm mitochondria implicated by complete resequencing of 41 genomes. BMC Evol Biol. 10:81.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the 9th Gateway Computing Environments Workshop (GCE), IEEE, 14 November 2010, New Orleans, LA; p. 1–8.
- Nagaraju J. 2000. Recent advances in molecular genetics of the silkmoth, Bombyx mori. Current Sci. 2:151–161.
- Park JS, Kim MJ, Jeong SY, Kim SS, Kim I. 2016. Complete mitochondrial genomes of two gelechioids, Mesophleps albilinella and Dichomeris ustalella (Lepidoptera: Gelechiidae), with a description of gene rearrangement in Lepidoptera. Curr Genet. 62:809–826.
- Ryu KS, Kim I, Ahn MY, Kim J-W, Lee P. 2003. Functionality research on silkworm and sericultural products. Food Sci Indus. 36:15–24.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Zhang H, Li F, Zhu X, Meng Z. 2016. The complete mitochondrial genome of Bombyx mori strain Baiyun (Lepidoptera: Bombycidae). Mitochondrial DNA Part A. 27:1652–1653.