
Abstract
In the present study, we determined the complete mitogenome sequence of scleractinia, Favia favus using the Illumina HiSeq platform. The assembled mitogenome was 17,054 bp in length, comprising unique 13 protein-coding genes (PCGs), 2 ribosomal RNAs, and 2 transfer RNAs genes of each, showing a typical scleractinian pattern. New phylogenetic analysis upon complete mitogenomics revealed that F. favus is most closely related to Favites abdita, with high bootstrap values.
In recent years, mass global bleaching event devastated the relatively healthy coral reefs (Roelfsema et al. Citation2018). Coral reefs, for instance, are increasingly under pressure due to coastal development and resource use (Shidqi et al. Citation2018). Mitochondria genome structure is important to the growth and developmental Progression in the Coral (Reyes-Bermudez et al. Citation2016). Favia favus, which belongs to the Faviinae family, is widely distributed throughout the Indo-Pacific. And it is most massive and spherical. The first establishment of mitogenome is important for further evolutionary and phylogenetic analyses for stony coral (Wang et al. Citation2018).
Samples (voucher no. YM03) of F. favus were collected from Yangmeiken in Guangdong, China (114°34′40.30″E, 22°32′47.63″N) on July 2018 and deposited at South China Sea Fisheries Research Institute Shenzhen test base Herbarium. We used the next-generation high-throughput sequencing method to acquire the F. favus complete mitochondrial genome sequences. The raw next-generation sequencing reads generated from HiSeq X-ten (Illumina, San Diego, CA). The reads were de novo assembly by using commercial software (SOAPdenovo V2.04, BGI, CHN) to produce a single, circular form of complete mitogenome with about an average 294 coverage. The complete mitogenome of F. favus was 17,054 bp in size (GenBank MK516277) and its overall base composition is 25.0% for A, 13.1% for C, 20.4% for G, and 41.5% for T, and have GC content of 33.5%, showing 100% identities to Favites abdita (GenBank KY094479.1). The protein coding, rRNA and tRNA genes of F. favus mitogenome were predicted by using DOGMA (DOGMA, the University of Texas at Austin, USA) (Wyman et al. Citation2004) server tools, and manually inspected. The complete mitogenome of F. favus includes unique 13 protein-coding genes (PCGs), 2 transfer RNA genes (tRNAMet, tRNA-Trp), and 2 ribosomal RNA genes. Among the 13 PCGs, the longest one is the ND5 gene (1815 bp), whereas the shortest is ATP8 gene (198 bp). The ND5 gene is split into two parts by a large fragment of genes, which commonly presented in scleractinian coral, and the size of the inserted fragment was usually over 10 Kb.
All PCGs, tRNA, and rRNA genes were encoded on H-strand. The most start codon is ATG. There are 10 PCGs started with ATG codon (ND1, ND3, ATP6, COX3, COX2, ND4, ND4L, ND5, ND6, and ATP8), two ATT codon (ND2 and COX1), and one with TAA codon (Cyt b). Nine of the 13 PCGs are inferred to terminate with TAA (ATP6, Cytb, ND3, ND2, ND6, ATP8, ND4L, COX2, and COX3), 4 with ATG (ND1, ND4, ND5, and COX1). The small and large mitochondrial ribosomal RNA genes of F. favus were located opposite each other on the circular genome as in other corals (Ju et al. Citation2017).
To determine the phylogenetic position of F. favus, we used MEGA Version10.0.4 (Knyaz et al. Citation2018) software to construct a Maximum likelihood tree using 500 bootstrap replicates and Kimura 2-parameter model (Tian and Niu Citation2017). The phylogenetic tree was reconstructed with 13 coral species complete mitogenomes derived from GenBank ().
Figure 1. Molecular phylogeny of Favia favus and related species in Hexacorallia based on complete mitogenome. The complete mitogenomes are downloaded from GenBank and the phylogenetic tree is constructed by maximum-likelihood method with 500 bootstrap replicates. The gene’s accession number for tree construction is listed behind the species name.
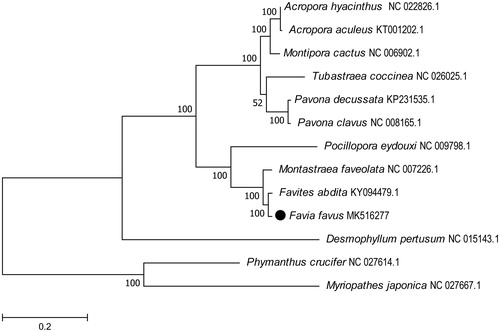
The result showed F. favus was grouped into a single clade of with Favites abdita. This relationship has been verified by molecular phylogenies. Which were with 100% bootstrap value supported. In conclusion, the complete mitogenome of the F. favus determined is expected to provide essential phylogenetic and evolutionary information of corals in this study.
The genus Favia, common across the Indo-Pacific, is characterized by a genus-specific septal formula. It is similar to genus Favites in appearance. Species identification to date has been conducted most using traditional macromorphologies, such as colony growth form, corallite dimensions, and number and fusion pattern of septa (Edinger and Risk Citation2000; Terraneo et al. Citation2016; Gonzálezespinosa et al. Citation2018). However, it is taxonomic uncertainty due to confusing patterns of morphological variation, with surprising examples of convergent evolution, rapid evolution, and phenotypic plasticity (Tisthammer and Richmond Citation2018).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Edinger EN, Risk MJ. 2000. Reef classification by coral morphology predicts coral reef conservation value. Biol Conserv. 92:1–13.
- Gonzálezespinosa PC, Pazgarcía DA, Reyesbonilla H, Cabraltena RA, Balart EF. 2018. Evidence of sexual dimorphism in skeletal morphology of a gonochoric reef coral. Royal Soc Op Sci. 5:1–7.
- Ju YM, Hsiao ST, Kuo FW, Wu JH. 2017. The complete mitochondrial genome of Montipora aequituberculata (Scleractinia, Acroporidae). Mitochondrial DNA Part B. 2:62–63.
- Knyaz C, Stecher G, Li M, Kumar S, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 35:1547–1549.
- Reyes-Bermudez A, Villar-Briones A, Ramirez-Portilla C, Hidaka M, Mikheyev AS. 2016. Developmental progression in the coral Acropora digitifera is controlled by differential expression of distinct regulatory gene networks. Genome Biol Evol. 8:851–870.
- Roelfsema C, Kovacs E, Ortiz JC, Wolff NH, Callaghan D, Wettle M, Ronan M, Hamylton SM, Mumby PJ, Phinn S. 2018. Coral reef habitat mapping: a combination of object-based image analysis and ecological modelling. Remote Sens Env. 208:27–41.
- Shidqi RA, Pamuji B, Sulistiantoro T, Risza M, Faozi AN, Muhammad AN, Muharam MR, Putri ED, Hartini R, Valentina B, et al. 2018. Coral health monitoring at Melinjo Island and Saktu Island: influence from Jakarta Bay. Region Stud Mar Sci. 18:237–242.
- Terraneo TI, Benzoni F, Arrigoni R, Berumen ML. 2016. Species delimitation in the coral genus Goniopora (Scleractinia, Poritidae) from the Saudi Arabian Red Sea. Mol Phylogenet Evol. 102:278–294.
- Tian P, Niu W. 2017. The complete mitochondrial genome of the Acropora pruinosa. Mitochondrial DNA Part B. 2:652–653.
- Tisthammer KH, Richmond RH. 2018. Corallite skeletal morphological variation in Hawaiian Porites lobata. Coral Reefs. 37:1–12.
- Wang X, Tian P, Niu W, Yu S. 2018. The complete mitochondrial genome of the Montipora peltiformi (Scleractinia: Acroporidae). Mitochondrial DNA Part B. 3:99–100.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.