
Abstract
Rosa acicularis Lindl. is a kind of ornamental species widely distributed in the north temperate zone. Here, we describe the complete chloroplast genome of R. acicularis using Illumina paired-end sequencing. The chloroplast genome length is 156,527 bp, including two inverted repeat (IR) region of 26,053 bp each, separated by a large single-copy (LSC) region of 85,674 bp and a small single-copy (SSC) region of 18,747 bp. The genome contains 129 genes, including 84 protein-codon genes (PCGs), 37 tRNA genes, and 8 rRNA genes. The whole chloroplast genome GC content is 37.2%. Phylogenetic analysis revealed that R. acicularis was closest related to R. praelucens.
Introduction
Rosa acicularis Lindl. is a shrub species, belonging to the family Rosaceae that originates from Heilongjiang, Jilin, Liaoning, Inner Mongolia, Hebei, Shaanxi, Shanxi, Gansu and Xinjiang, at altitudes of 400–1800 m. It is also found in Japan, Kazakhstan, Korea, Mongolia, Russia, Northern Europe and North America. It has a great morphological variation. The florescence of R. acicularis is from June to July, it has ornamental value. However, its chloroplast genome has not been reported. In this study, we assembled the complete chloroplast (cp) genome sequence of R. acicularis.
Sample of R. acicularis was collected from Genhe province (Inner Mongolia, China; 121°36′1.00″ E, 50°50′44.00″ N). The specimen voucher number is BOP011170. Genomic DNA was extracted from the dried leaves using the mCTAB (modified cetyltrimethyl ammonium bromide) approach (Li et al. Citation2013). The whole genome sequences were obtained using Illumina Hiseq PE150 Platform (Illumina, San Diego, CA). The chloroplast genome data were extracted using R. multiflora (MG893867) as a reference and assembled de novo with SPAdes and reconfirmed with Geneious (Bankevich et al. Citation2012; Kearse et al. Citation2012). The initial annotation was performed on Plann and correct with Sequin (Huang and Cronk Citation2015). The data were compared homologous genes in R. multiflora to determine the putative starts, stops, and intron positions. The complete cp genome sequence with gene annotations were submitted to GenBank under the accession number of MK714016 for R. acicularis.
The complete cp genome of R. acicularis with a size of 156,527 bp in length is a circular molecular genome, including two inverted repeat (IR) region of 26,053 bp each, separated by a large single-copy (LSC) region of 85,674 bp, and a small single-copy (SSC) region of 18,747 bp. In total, the genome contained 129 genes, including 84 protein-codon genes (PCGs), 37 tRNA genes, and 8 rRNA genes. Among them, 16 genes occur in double copies, including 5 protein-codon genes (PCGs), 7 tRNA genes, and 4 rRNA genes, and most of the gene occur as a single copy. The whole chloroplast genome GC content is 37.2%, while the value of the LSC, SSC, and IR regions are 35.2%, 31.3%, and 42.7% respectively.
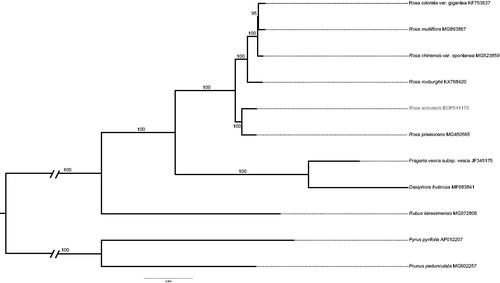
We used the plastid genomes of other 10 Rosaceae species, including five species of Rosa, five species of Rosoideae or Amygdaloideae (outgroup), to validate the phylogenetic position of R. acicularis. The sequences were aligned by MAFFT version 7 (Katoh and Standley Citation2013). ModelFinder (Kalyaanamoorthy et al. Citation2017) was used for model selection according to the Bayesian information criterion (BIC), and a maximum likelihood (ML) tree with 1000 bootstrap replicates were inferred by IQ-TREE (Nguyen et al. Citation2015). The phylogenetic result indicates that R. acicularis is most closely related to R. praelucens (100% ultrafast bootstrap support) (Hoang et al. Citation2018) (). The complete cp genome of R. acicularis can lay the foundation for phylogenetic analyses, cp genetic engineering studies, and population genomic studies of Rosaceae.
Figure 1. Phylogenetic tree based on plastid genomes using the ML method. Ultrafast bootstrap (UFBoot) values are shown above the nodes, with 1000 bootstrap replicates.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35:518–522.
- Huang DI, Cronk QCB. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3:1500026.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods. 14:587.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Li JL, Wang S, Yu J, Wang L, Zhou SL. 2013. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 48:72–78.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.