
Abstract
The European beech (Fagus sylvatica) is one of the most important and widespread tree species in Central Europe and is widely managed for its valuable hard wood. We assembled and characterized the complete chloroplast genome of F. sylvatica to serve as a valuable resource in future genetic studies. The complete plastome sequence is 158,462 bp in length and contains 131 genes including 83 protein-coding genes, 40 tRNA genes, and 8 rRNA genes. A phylogenetic analysis of 21 Fagaceae plastome sequences shows that the three available Fagus sequences form a cluster, suggesting that the genus Fagus represents a monophyletic clade within the Fagaceae.
Fagus is a genus of deciduous trees in the family Fagaceae, native to temperate Europe, Asia, and North America. The genus is divided into two subgenera, Engleriana and Fagus (Renner et al. Citation2016). As a naturally growing forest tree, European beech (F. sylvatica L.; subgenus Fagus) is one of the most important and widespread tree species in Central Europe and is widely managed for its valuable hard wood. Despite the significance of the genus Fagus, complete chloroplast genomes of only two species are so far publicly available (F. engleriana (Yang et al. Citation2018); F. crenata (Worth et al. Citation2019)). This study reports the whole plastome sequence of F. sylvatica, thus extending genomic sequence resources for this species (nuclear scaffolds; (Mishra et al. Citation2018)).
The reference tree (FASYL_29; population 46, plot1, tree No. 12) is located at the 25 years old provenance trial Dobersdorf, Schädtbek, Germany (54°18′ N, 10°18′ E, 40 m a.s.l.). The parental population (original provenance) of the tree is the German population Gransee/Brandenburg, located in the center of the natural range (53°00′ N, 13°10′ E, 70 m a.s.l.). The selected individual is a good genetic representative of Central Europe based on a former genotyping study (Liesebach et al. Citation2015). Dormant buds of the reference tree were sampled, green tissues were dissected and total DNA (Voucher specimen: sample accession FASYL_29_1; stored at the Thünen Institute) was extracted according to the method described by Dumolin et al. (Citation1995).
Standard genomic library preparation and 300 bp paired end-sequencing was performed on Illumina MiSeq at 24× coverage (GATC Biotech AG, Konstanz, Germany). Reads were trimmed with Trimmomatic (Bolger et al. Citation2014) and assembled using the CLC Genomics Workbench (CLC-GWB) Version 10.1.1 (CLC-bio, a Qiagen company; Aarhus, Denmark), (Length fraction = 0.9, Similarity fraction = 0.95, Map reads back to contigs, Word size = 45). Three out of 279,689 contigs were identified as chloroplast sequences based on high mapping coverage and comparison against the NCBI nucleotide collection database. Blastn was used to join the chloroplast contigs by finding similarities between the sequence endings. The annotation of F. engleriana was used as a template to annotate the resulting circular sequence of F. sylvatica using the tools GB2Sequin (Lehwark and Greiner Citation2018) and Sequin (Sequin – A DNA Sequence Submission Tool Citation2019).
The complete chloroplast genome sequence of F. sylvatica (MK598696) has a total length of 158,462 bp and consists of a large single-copy region (87,700 bp), a small single-copy region (19,010 bp), and two inverted repeat regions (25,870 bp, each). The annotated sequence contains 131 genes including 83 protein-coding genes, 40 tRNA genes, and 8 rRNA genes. The CG content of the complete sequence averages 37.1%.
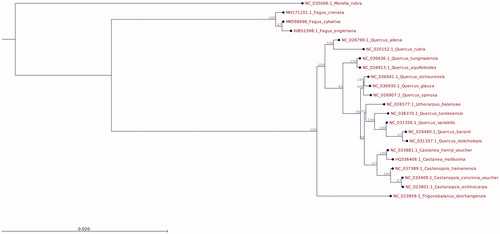
A phylogenetic tree was created based on a multiple sequence alignment (CLC-GWB Version 12) of 21 complete chloroplast DNA sequences of Fagaceae species including F. sylvatica, F. engleriana and F. crenata. Morella rubra (Fagales, Myricaceae) was used as an out-group. The tree indicates that the genus Fagus forms a monophyletic clade within the Fagaceae ().
Figure 1. Phylogenetic tree (maximum likelihood) based on whole-plastome sequences of Fagaceae species and Morella rubra (outgroup). Bootstrap values (%) are shown above branches. The phylogenetic tree was constructed based on a whole-plastome alignment using the ‘Maximum likelihood phylogeny’ tool of CLC-GWB including bootstrap analysis with 100 replicates (other parameters: construction method for the start tree = UPGMA; existing start tree = not set; nucleotide substitution model = Jukes Cantor; protein substitution model = WAG; transition/transversion ratio = 2.0; include rate variation = No; number of substitution rate categories = 4; gamma distribution parameter = 1.0; estimate substitution rate parameter(s) = Yes; estimate topology = Yes; estimate gamma distribution parameter = no).
Acknowledgements
We would like to thank Vivian Kuhlenkamp for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England). 30:2114–2120.
- Dumolin S, Demesure B, Petit RJ. 1995. Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet. 91:1253–1256.
- Lehwark P, Greiner S. 2018. GB2sequin – a file converter preparing custom GenBank files for database submission. Genomics. S0888-7543(0818)30189-30187.
- Liesebach H, Eusemann P, Liesebach M. 2015. Verwandtschaftsbeziehungen innerhalb von Prüfgliedern in Herkunftsversuchen – Beispiel Buche (Fagus sylvatica L.) [Sibship structure in samples from a provenance trial. A case study in beech (Fagus sylvatica L. )]. Forstarchiv. 86:174–182.
- Mishra B, Gupta DK, Pfenninger M, Hickler T, Langer E, Nam B, Paule J, Sharma R, Ulaszewski B, Warmbier J, et al. 2018. A reference genome of the European beech (Fagus sylvatica L.). GigaScience. 7:giy063.
- Renner SS, Grimm GW, Kapli P, Denk T. 2016. Species relationships and divergence times in beeches: new insights from the inclusion of 53 young and old fossils in a birth–death clock model. Phil Trans R Soc B. 371:20150135.
- Sequin – a DNA sequence submission tool. 2019. [accessed 01/02/2019]. https://www.ncbi.nlm.nih.gov/Sequin/.
- Worth J, Liu L, Tomaru N. 2019. The complete chloroplast genome of Fagus crenata (subgenus Fagus) and comparison with F. engleriana (subgenus Engleriana). PeerJ. 7:e27473v1.
- Yang Y, Zhu J, Feng L, Zhou T, Bai G, Yang J, Zhao G. 2018. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Front Plant Sci. 9:82.