
Abstract
Mitochondrial genomes have proved to be a powerful tool for studying evolutionary relationships and the evolutionary history. In this study, the complete mitochondrial genome of Emberiza pallasi (16,786 bp) was determined. It is similar in architecture to the typical vertebrate mtDNA genome, which consists of 37 genes (13 protein-coding genes, 2 ribosomal RNA genes, and 22 transfer RNA genes) and one control region. Overall base composition of the mtDNA was A (29.37%), T (22.65%), C (33.23%), and G (14.75%). Phylogenetic analysis based on Emberiza mitogenomes indicated a relative close relationship among E. pallasi with E. elegans and E. siemsseni.
The Pallas’s bunting, Emberiza pallasi (Passeriformes, Emberizidae, Emberiza), is a small songbird, which is similar to a reed bunting (E. schoeniclus). The male has a black head, chest and throat, white neck, and a heavily streaked grey back (while the reed bunting has a browner back). The female has a black-brown head and is less streaked below than the female reed bunting. It breeds in northern and central Asia across to Mongolia (Gill and Donsker Citation2018). The E. pallasi used in this study was collected from Dashu Mountain, Hefei, Anhui Province, China (November, 2016) and stored in the museum of Anhui Key Laboratory of Eco-engineering and Bio-technique, Anhui University, China.
Mitochondrial DNA (mtDNA) has proven to be a powerful tool for taxonomy, phylogenetic relationships, phylogeography and evolutionary history (Boore Citation1999; Van et al. Citation2000; Galtier et al. Citation2009; Li et al. Citation2015). In this study, total genomic DNA of E. pallasi was extracted using a standard proteinase K/phenol-chloroform protocol (Sambrook et al. Citation1982). The complete mtDNA sequence was amplified using 16 pairs of primers and submitted into GeneBank (accession number MK687386). The complete mtDNA sequence of the E. pallasi genome is 16,786 bp in length, consisting of 13 protein-coding genes, 22 tRNA genes, 2 rRNA genes, and a control region (D-loop region). In 13 protein-coding genes, the shortest one is ATP8 gene (168 bp), and the longest one is the ND5 gene (1818 bp). As in most vertebrates, all of the genes in E. pallasi were distributed on the H-strand, except the ND6 subunit and eight tRNA genes, which were encoded on the L-stand. The overall base composition of the mtDNA was A (29.37%), T (22.65%), C (33.23%), and G (14.75%), the percentage of A and T (52.02%) being slightly higher than that of G and C (47.98%).
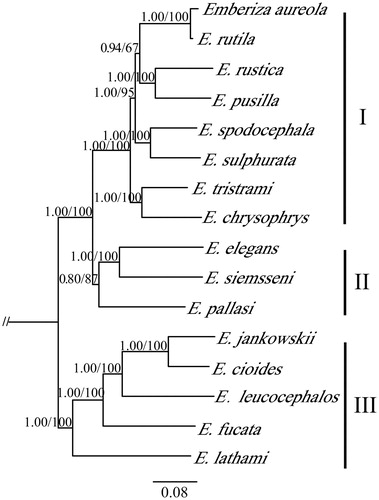
Phylogenetic trees were estimated based on the mitochondrial genome dataset of 16 Emberiza species and out-group taxa using Bayesian inference (BI) and maximum likelihood (ML) methods, implemented in MrBayes 3.2.2 (Ronquist et al. Citation2012) and RaxML version 8 (Stamatakis Citation2014), respectively. The result showed that the Emberiza divided into three major clades (). The E. pallasi located in a separated small branch with E. elegans and E. siemsseni. It presented a relative close relationship with E. elegans and E. siemsseni at the molecular level. We expect the complete mitogenome of E. pallasi to provide a useful data for further study of Emberiza species.
Figure 1. Phylogenetic tree of Emberiza species based on the mitogenome. The values on nodes indicate the Bayesian posterior probabilities and ML bootstrap proportions calculated. GenBank accession numbers: E. aureola, NC022150; E. chrysophrys, HQ896034; E. cioides, NC024524; E.elegans, NC030368; E. fucata, KT737824; E. jankowskii, NC027251; E. leucocephalos, KU356573; E. lathami, NC031845; E. pallasi, MK687386; E. pusilla, NC021408; E. rustica, NC024924; E. rutile, NC024925; E. siemsseni, NC032304; E. spodocephala, NC021445; E. sulphurata, KY419885; E. tristrami, NC015234.
Acknowledgements
We sincerely thank Zhaojie Peng, Lifu Qian, Chencheng Wang, Ke Fang and Xing Kang for their help in sample collection and Lab work.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27:1767–1780.
- Galtier N, Nabholz B, Glémin S, Hurst G. 2009. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Mol Ecol. 18:4541–4550.
- Gill F and Donsker D (Eds). 2018. IOC World Bird List (v 8.2). http://www.worldbirdnames.org/ (accessed on Dec. 7th, 2018).
- Li X, Huang Y, Lei F. 2015. Comparative mitochondrial genomics and phylogenetic relationships of the Crossoptilon species (Phasianidae, Galliformes). BMC Genomics. 16:42.
- Ronquist F, Teslenko M, Van dMP, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. System Biol. 61:539–542.
- Sambrook J, Fritsch EF, Maniatis T. 1982. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor laboratory press.
- Stamatakis A. 2014. RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Van TM, Sibley CG, Hedges SB. 2000. The early history of modern birds inferred from DNA sequences of nuclear and mitochondrial ribosomal genes. Mol Biol Evol. 17:451–457.