
Abstract
Eryx is a genus of snakes belonging to the family Boidae. In this study, the mitochondrial genome sequence of Eryx tataricus was generated using a PacBio RSII DNA sequencer employing the single molecule, real-time sequencing technology. A maximum-likelihood (ML) phylogenetic tree of 26 snakes was re-constructed based on the 13 protein-coding genes for convincing the mitochondrial DNA sequences.
Eryx tataricus (Lichtenstein, 1823) is a reprehensive species of genus Eryx which is a group of nonvenomous boas and has a wide distribution in world. Eryx tataricus is mainly distributed in desert regions. However, with the accelerating development of tourism and devastating poaching, this species displays a marked population degradation. The complete sequence of mitochondrial genome could provide plenty of information for species identification, population history inference, as well as phylogenetic analysis (e.g. Imoto et al. Citation2013). In this study, the complete mitochondrial genome of E. tataricus was assembled and characterized.
The specimens of E. tataricus were collected from Xiaoguai village, Karamay, Xinjiang province, China (longitude: 85°3′5.19″, latitude: 45°10′10.74″), and deposited at CIB herpetological museum, Chengdu Institute of Biology with the number CIB116071. The genome sequences were generated by the PacBio RSII DNA sequencer and reserved in the GenBank with the accession number of MK780743. Eryx tataricus mitochondrial genome was compared with Boa constrictor which was downloaded from the GenBank (AB177354.1) as a reference sequence. The annotation of the E. tataricus mitochondrial genome were firstly generated with the MITOS WebServer (Bernt et al. Citation2013) and then with manual correction. All the structures of tRNA genes were analyzed using tRNAScan-SE (Lowe and Chan Citation2016).
For phylogenetic analyses of E. tataricus, IQTREE v1.6.10 (Nguyen et al. Citation2015) was used to reconstruct a maximum-likelihood (ML) tree with 13 protein-coding genes from 26 snakes. The alignment was performed using PRANK v150803 (Loytynoja and Goldman Citation2005) with options ‘–F– codon’. All gaps and poorly aligned positions were removed using Gblocks v0.91b (Castresana Citation2000). ModelFinder (Kalyaanamoorthy et al. Citation2017) was used to find the best fitting substitution model (GTR + F+R4) for phylogenetic inference.
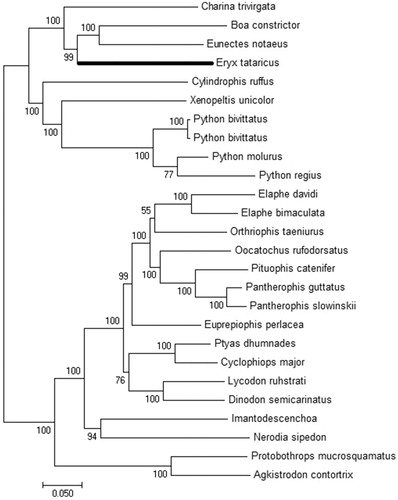
As a result, the mitogenome sequence of E. tataricus consists of 19,330 bp length and contains two ribosomal RNA genes (12s and 16s rRNA), 13 protein-coding genes (PCGs), and 22 transfer RNA (tRNA) genes while the tRNA-Leu and tRNA-Ser were repeated detected. The overall base composition was 37.7% for A, 26.2% for T, 24.3% for C, and 11.9% for G. All protein-coding genes were encoded on the H-strand with exception of ND6. The sequence lengths of 12s and 16s rRNA were 922 bp and 1517 bp, respectively, which were separated by tRNA-Val. Our predictive annotation resulted in the canonical ATN start codon for 10 genes. COX1, COX2, and ND5 had the GTG start codon. The typical T++ stop codons are present in PCGs except for ND4L and COX3. The obtained phylogenetic tree () strongly indicated that E. tataricus has the close relationship with B. constrictor and Eunectes notaeus. This resource is the first complete mitochondrial genome of E. tataricus that is generated through de novo next-generation sequencing and will provide a reliable reference for further comparative mitogenome studies as well as the evolutionary relationships within Boidae families.
Figure 1. Phylogenetic tree of the relationships among snakes based on maximum likelihood (ML) method. The number nearby the node is the bootstrap supporting value. The species selected and corresponding GenBank accession number are shown as follows: Eryx tataricus (MK780743), Eunectes notaeus (AM236347), Charina trivirgata (GQ200595), Boa constrictor (AB177354), Python bivittatus (KF010492), Python bivittatus (KF293729), Python regius (AB177878), Python molurus molurus (HM581978), Ptyas dhumnades (KF148621), Imantodes cenchoa (EU728586), Protobothrops mucrosquamatus (KC438281), Cylindrophis ruffus (AB179619), Lycodon ruhstrati (KJ179951), Oocatochus rufodorsatus (KC990020), Orthriophis taeniurus (KC990021), Dinodon semicarinatus (AB008539), Nerodia sipedon (JF964960), Xenopeltis unicolor (AB179620), Agkistrodon contortrix (KY747498), Cyclophiops major (KF148620), Pantherophis guttatus guttatus (AM236349), Elaphe davidi (KM401547), Elaphe bimaculata (KM065513), Pantherophis slowinskii (DQ523162), Euprepiophis perlacea (KF750656), and Pituophis catenifer sayi (KU833245).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552.
- Imoto JM, Saitoh K, Sasaki T, Yonezawa T, Adachi J, Kartavtsev YP, Miya M, Nishida M, Hanzawa N. 2013. Phylogeny and biogeography of highly diverged freshwater fish species (Leuciscinae, Cyprinidae, Teleostei) inferred from mitochondrial genome analysis. Gene. 514:112–124.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14:587–589.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44:W54–57.
- Loytynoja A, Goldman N. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA. 102:10557–10562.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.