
Abstract
Trichoplusia ni is a polyphagous insect that feeds on many crops plants of economic importance. In this paper, we presented the complete mitochondrial genome of T. ni. The circular mitochondrial genome size is 15,239 bp, containing 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNA), two ribosome RNA genes (rRNA), and a control region of 1103 bp in length. The base composition of the genome is as follows: A of 38.9%, T of 41.6%, C of 11.5%, and G of 8.0%, with a total G + C content of the mitochondrial genome 19.5% and A + T of 80.5%. The phylogenetic Neighbour-Joining (NJ) tree was constructed to validate the taxonomic status of T. ni, exhibiting a closest relationship with Spodoptera litura and Spodoptera frugiperda. The research of the complete mitochondrial genome of Trichoplusia ni is able to provide a reference taxonomic relationships and more data of the insect for further.
Cabbage looper, Trichoplusia ni, is a polyphagous insect that feeds over 160 species of plants and crops of economic importance, such as cruciferous plants with a preference for cabbage. It is found throughout in the world, the southern Palaearctic ecozone, including all of North America, parts of Africa, much of eastern Europe and the Indo-Australian region (Franklin et al. Citation2011). This species is very destructive to plants due to its voracious consumption of foliage (Akhtar et al. Citation2009, Citation2010). Cabbage looper has evolved resistance against many synthetic insecticides and the microbial insecticide that it is important to develop new method that could be used to protect crops at present (Tak and Isman Citation2015). Now, there are no studies of T. ni genome, also little knowledge about the mitochondrial genome of this species. So, in this study, we provide the complete mitochondrial genome of T. ni, which constitutes a valuable and useful resource for population genetic and genetic diversity study conservation in China.
The specimen sample of T. ni was collected and deposited from Yu ling (Yuling, Shaanxi, China, 109.77E; 38.30N). The whole insect tissue and total genomic DNA of T. ni were extracted using Insect Tissues Genomic DNA Extraction Kit (Bioer, HZ, CN) and stored in Yulin University College of Life Science. The whole genomic DNA was purified and fragmented using the NEB Next Ultra™ II DNA Library Prep Kit (NEB, BJ, CN) that the whole genomic was sequenced using the Illumina HiSeq 4000 Sequencing Platform (Illumina, San Diego, CA). Quality reads and adapters control was performed and removed low-quality reads and adapters using the NGS QC Toolkit (Patel and Jain Citation2012). The mitochondrial genome was assembled and annotated using the MitoZ (Meng et al. Citation2019). Protein-coding genes were predicted using the mitochondrial genetic codes on the MITOS web server (Matthias et al. Citation2013). The physical map of the new mitochondrial genome was generated using OGDRAW (Lohse et al. Citation2013).
The complete mitochondrial genome of T. ni (GenBank accession No. MK714850.1) was a closed-circle with 15,239 bp in length, which is well within the length range observed in the sequenced mitogenomes. The mtDNA of T. ni comprised 37 genes, including 13 protein-coding genes (PCG) (ND1-6, ND4L, ATP6, ATP8, COX1-3, and CytB), 22 transfer RNA genes (tRNA) (one for each amino acid, two each for Serine, and Leucine amino acid), two ribosomal RNA genes (rRNA) (12S rrn and 16S rrn) and a control region that is 1103 bp in size. The base composition of the genome is as follows: 38.9% A, 41.6% T, 11.5% C, and 8.0% G, with a total GC content of 19.5% and AT of 80.5%.
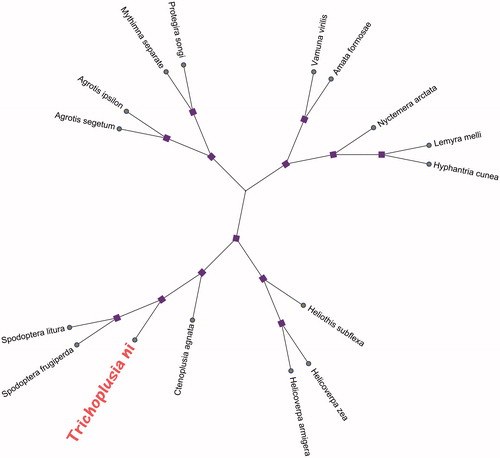
To elucidate the phylogenetic position of T. ni, the phylogenetic tree was constructed with 15 published complete mitochondrial genomes of 13 PCGs with T. ni. Phylogenetic analysis using the Neighbour-Joining (NJ) methods, we selected the protein-coding amino acid sequences were aligned using the program MAFFT version 5.0 and adjusted manually (Katoh et al. Citation2005). NJ methods analysis was performed using MEGA X (Kumar et al. Citation2018) with the number of bootstrap replicates with 10,000. All of the nodes were inferred with strong support by the NJ methods. The phylogenetic NJ tree was represented using MEGA X (Kumar et al. Citation2018) and edited using iTOL web server (https://itol.embl.de/). The phylogenetic NJ tree based on the protein-coding amino acid sequences of 13 PCGs with excluding the stop codons as shown in , the mitochondrial genome of T. ni taxonomic status was closest with Spodoptera litura (GenBank accession No. NC_022676.1) and Spodoptera frugiperda (GenBank accession No. KM362176.1). The complete mitochondrial genome sequence provides more molecular data for the genetic diversity conservation of this species.
Figure 1. The phylogenetic relationships of 16 species mitochondrial genomes based on concatenated amino acid sequences of the 13 PCGs. Numbers above each node indicates the NJ bootstrap support values. Fifteen species accession numbers in this study have been deposited in the GenBank are as follows: Agrotis ipsilon (NC_022185.1), Agrotis segetum (NC_022689.1), Amata formosae (NC_021416.1), Ctenoplusia agnate (NC_021410.1), Helicoverpa armigera (NC_014668.1), Helicoverpa zea (NC_030370.1), Heliothis subflexa (NC_028539.1), Hyphantria cunea (NC_014058.1), Lemyra melli (NC_026692.1), Mythimna separate (NC_023118.1), Nyctemera arctata (KM244681.1), Protegira songi (NC_034938.1), Spodoptera litura (NC_022676.1), Spodoptera frugiperda (KM362176.1), and Vamuna virilis (NC_026844.1).
Acknowledgments
The authors thank Dr. Xiongfei Yan for providing the whole genomic DNA of Trichoplusia ni.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Akhtar Y, Shikano I, Isman MB. 2009. Topical application of a plant extract to different life stages of Trichoplusia ni fails to influence feeding or oviposition behaviour. Entomol Exp Appl. 132:275–282.
- Akhtar Y, Yu Y, Isman MB, Plettner E. 2010. Dialkoxybenzene and dialkoxyallylbenzene feeding and oviposition deterrents against the cabbage looper, Trichoplusia ni: potential insect behavior control agents. J Agric Food Chem. 58:4983–4991.
- Franklin MT, Ritland CE, Myers JH. 2011. Genetic analysis of cabbage loopers, Trichoplusia ni (Lepidoptera: Noctuidae), a seasonal migrant in western North America. Evol Appl. 4:89–99.
- Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33:511–518.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:W575–W581.
- Matthias B, Alexander D, Frank J, Fabian E, Catherine F, Guido F, Joern P, Martin M, Peter FS. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Meng G, Li Y, Yang C, and, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. gkz173.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7:e30619.
- Tak JH, Isman MB. 2015. Enhanced cuticular penetration as the mechanism for synergy of insecticidal constituents of rosemary essential oil in Trichoplusia ni. Sci Rep. 5:12690.