
Abstract
The complete mitochondrial genome was determined for a whitefly (Hemiptera: Aleyrodidae) infesting soybean Glycine max (hereafter whitefly_Glycine max_China) (GenBank accession number: MK645228). The mitochondrial genome has a total length of 15,074 bp and contains 13 protein-coding genes (PCGs), 22 transfer RNAs, and two ribosomal RNAs. The overall base composition is 29.7% A, 42.4% T, 15.9% G, and 12.0% C, with an A + T bias of 72.1%. All PCGs start with ATN codon, and all PCGs end with TAA codon except COX1, COX2, and NAD5. The resultant maximum-likelihood and Bayesian inference trees based on 13 PCGs support a close relationship of this whitefly with Tetraleurodes acaciae. Gene arrangement of the 13 PCGs between the whitefly_Glycine max_China and T. acaciae is identical and the COX1 sequences of the two species share a similarity of 78.95%.
Whiteflies (Hemiptera: Aleyrodidae) comprise at least 1556 species in 161 genera (Martin and Mound Citation2007). Some of them are often found infesting economic plants around the world (Martin Citation1987). The classification of whiteflies is based on the characteristics of the fourth instar nymphs. However, their characteristics may vary with host plants and possibly other factors (Mound Citation1963; Jesudasan and David Citation1991). Molecular marker is a valuable tool for aiding species identification and mitochondrial genome sequence provides complementary evidence for the classification and phylogenetic analysis of whiteflies (Wang et al. Citation2016).
This article reports the complete mitochondrial genome of a whitefly infesting soybean Glycine max (hereafter whitefly_Glycine max_China). The specimens were collected from Jiangxi, China (29°43′42″N, 115°47′32″E) and deposited at the Institute of Insect Sciences, Zhejiang University, Hangzhou, China. Total genomic DNA was extracted from individual adults with DNeasy Blood & Tissue Kit (Qiagen, Germany) and next-generation sequencing was performed with Illumina HiSeq 4000 platform (2*150 bp) at Shanghai Biozeron Biotech. Co., Ltd. Raw reads were filtered and assembled using ABySS (Jackman et al. Citation2017) and GapCloser (Luo et al. Citation2012) with available whitefly mitochondrial genomes as references. The assembled mitochondrial genome sequence was annotated with DOGMA (Wyman et al. Citation2004) and tRNAscan-SE (Lowe and Eddy Citation1997).
The complete mitochondrial genome of the whitefly_Glycine max_China is 15,074 bp in length (GenBank accession number: MK645228) and contains 13 protein-coding genes (PCGs), 22 tRNAs, and two rRNAs. The overall base composition is 29.7% A, 42.4% T, 15.9% G, and 12.0% C, with an A + T bias of 72.1%. All PCGs use ATN as start codon, and all PCGs use TAA as stop codon except for three genes where COX1 ends with TAG and both COX2 and NAD5 end with a single T. Gene arrangement of the 13 PCGs is identical to that of Aleurocanthus camelliae (KU761949), Aleurocanthus spiniferus (NC_029155), Bemisia afer (NC_024056), Bemisia tabaci (JQ906700), Neomaskellia andropogonis (NC_006159) and Tetraleurodes acaciae (AY521262), while differs from that of Aleurochiton aceris (AY572538), Aleurodicus dugesii (AY521251), Trialeurodes vaporariorum (AY521265), and whitefly_Litchi chinensis_China (MH999477), respectively.
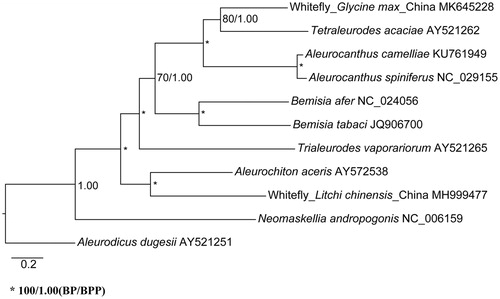
Using A. dugesii (subfamily Aleurodicinae) as an outgroup, the phylogeny of nine Aleyrodinae species and whitefly_Glycine max_China was analysed based on nucleotide sequence of 13 PCGs. The sequences were aligned using multiple sequence alignment program ClustalW in software MEGA v7.0 (Kumar et al. Citation2016). All gaps and poorly aligned positions were removed using Gblocks (Talavera and Castresana Citation2007). The phylogenetic relationships were reconstructed using the maximum-likelihood and Bayesian inference methods through RAxML (Stamatakis Citation2006) and MrBayes v3.2.6 (Ronquist et al. Citation2012). The resultant maximum-likelihood and Bayesian inference trees support that the whitefly_Glycine max_China belongs to the subfamily Aleyrodinae and is closely related to T. acaciae (). Additionally, the whitefly_Glycine max_China COX1 sequence is 78.95% similar to that of T. acaciae.
Figure 1. Maximum likelihood and Bayesian inference phylogenetic trees inferred from the nucleotide sequence of mitogenomic 13 PCGs.
Disclosure statement
The authors report no conflicts of interest and are alone responsible for the content and writing of this paper.
Additional information
Funding
References
- Jackman SD, Vandervalk BP, Mohamadi H, Chu J, Yeo S, Hammond SA, Jahesh G, Khan H, Coombe L, Warren RL, et al. 2017. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res. 27:768–777.
- Jesudasan RWA, David BV. 1991. Taxonomic studies on Indian Aleyrodidae (Insecta: Homoptera). Orient Insects. 25:231–434.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33:1870–1874.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 1:18.
- Martin JH. 1987. An identification guide to common whitefly pest species of the world (Homopt Aleyrodidae). Trop Pest Managem. 33:298–322.
- Martin JH, Mound LA. 2007. An annotated check list of the world's whiteflies (Insecta: Hemiptera: Aleyrodidae). Zootaxa. 1492:1–84.
- Mound LA. 1963. Host correlated variation in Bemisia tabaci (Gennadius) (Homoptera: Aleyrodidae). Proc R Ent Soc Lond (A). 38:171–180.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22:2688–2690.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56:564–577.
- Wang HL, Zhang Z, Bing XL, Liu YQ, Liu SS, Wang XW. 2016. A complete mitochondrial DNA genome derived from a Chinese population of the Bemisia afer species complex (Hemiptera: Aleyrodidae). Mitochondrial DNA A. 27:3500–3501.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20:3252–3255.