
Abstract
Sweet cherry (Prunus avium L.) is one of the two major species of cherries in world trade. In this study, we determined the complete mitochondrial genome of sweet cherry cultivar ‘summit’ using whole genome sequencing data. The genome was 389,709 bp in size. Totally, 44 genes were predicted, including 25 protein-coding, 16 tRNA, and 3 rRNA genes. A maximum-likelihood phylogenetic tree was reconstructed based on the complete mitochondrial genome sequences of eight species within Rosales. The result indicated that P. avium was clustered together with other Rosaceae species with high support value. The complete mitochondrial genome sequence of P. avium could provide valuable information for understanding the phylogenetic relationships within the Rosaceae family.
Sweet cherry (Prunus avium L.) is one of the most important fruit crops of the Rosaceae family. It is distributed widely in the areas of the world with a temperate climate (Kappel et al. Citation2012). World production of sweet cherries has been increasing steadily during the last two decades (Chen et al. Citation2018). It is reported that more than 2.8 million tons of sweet cherry fruit were produced in 2014 (Piaskowski et al. Citation2018). Much of the increase in production is taken up by new cultivars, many developed by fruit breeding programs from around the world (Kappel et al. Citation2012). As powerful tools for fruit breeding programs (Yuan et al. Citation2018), genomic resources for sweet cherry like nuclear and chloroplast genome sequence have been released (Shirasawa et al. Citation2017; Chen et al. Citation2018). However, little is known about the complete mitochondrial genome of sweet cherry.
In our present study, the complete mitochondrial genome of sweet cherry cultivar ‘summit’ was determined using the whole genome sequencing data. Total genomic DNA was extracted from mature leaves collected from the experimental orchard of Nanjing Forestry University (116.355 E, 40.013 N) using a modified CTAB protocol. The voucher specimen was deposited at Nanjing Forestry University. Qualified DNA was fragmented and used to prepare paired-end libraries following the Illumina DNA manufacturer’s instructions. 2 × 150 bp paired-end libraries with an average inserted-size 400 bp were sequenced on Illumina Hiseq X Ten platform (Illumina, San Diego, CA). Reads of mitochondrial origin were selected using GetOrganelle (Jin et al. Citation2018) with the mitochondrial genome sequence of Malus domestica as reference (Goremykin et al. Citation2012). All selected paired-end reads were used to assemble a complete mitochondrial genome using NOVOPlasty (Dierckxsens et al. Citation2017). Genome annotation was performed with the online program GeSeq (Tillich et al. Citation2017). The result was inspected and adjusted manually where necessary using Geneious. The annotated mitochondrial genome of sweet cherry has been submitted to Genbank with accession number MK816392.
The complete mitochondrial genome is 389,709 bp in length with an overall GC content of 45.62%. A total of 44 genes were found, including 25 protein-coding, 3 rRNA, and 16 tRNA genes. Five genes (nad1, nad2, nad4, nad5, nad7) have exon-intron structure and three protein-coding genes (nad1, nad2, nad5) are trans-spliced. The protein-coding genes in the sweet cherry mitochondrial genome account for 5.8% of the genome and a total length of 22,765 bp, while tRNA genes and rRNA genes only represent 1,194 and 5,134 bp of the genome, respectively. Most of the protein-coding genes use the common start codon: ATG, except nad1, which uses GTG as a start codon.
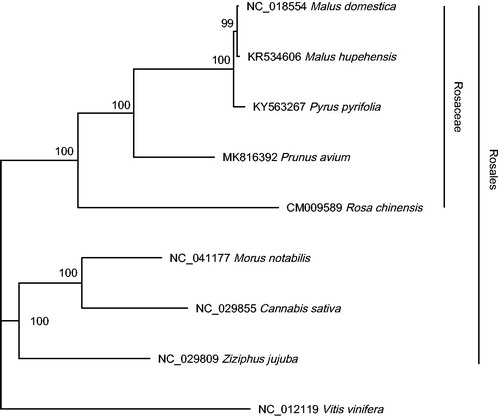
Phylogenetic analysis was performed using the complete mitochondrial genomes of eight species within Rosales. Vitis vinifera was selected as outgroup. Multiple sequences alignment was achieved by HomBlocks pipeline (Bi et al. Citation2018). Maximum-likelihood analysis was conducted using IQ-tree (Nguyen et al. Citation2015) with 1,000 replicates of ultrafast bootstrapping. The best-fit model was selected using ModelFinder (Kalyaanamoorthy et al. Citation2017). The result () showed that P. avium formed a single clade with other Rosaceae species. The complete mitochondrial genome of P. avium will provide valuable information for understanding the phylogenetic relationships within Rosaceae.
Figure 1. Phylogenetic tree of Prunus avium cv. ‘summit’ with other eight species belonging to Rosales. Tree was inferred from the complete mitochondrial genome sequences using the maximum likelihood method. Numbers in the nodes represent the bootstrap values from 1,000 replicates.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110:18–22.
- Chen T, Hu G, Wang Y, Chen Q, Wang L, Zhang J, Tang H, Wang X. 2018. Characterization of complete chloroplast genome and phylogenetic analysis of sweet cherry Cerasus avium (L.) Moench (Prunoideae, Rosaceae). Mitochondrial DNA Part B. 3:1274–1275.
- Chen T, Wang Y, Wang L, Chen Q, Zhang J, Tang H, Wang X. 2018. The complete chloroplast genome of Tomentosa cherry Prunus tomentosa (Prunoideae, Rosaceae). Mitochondrial DNA Part B. 3:672–673.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Goremykin VV, Lockhart PJ, Viola R, Velasco R. 2012. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71:615–626.
- Jin J, Yu W, Yang J, Song Y, Yi T, Li D. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. bioRxiv. 25679.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler AV, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14:587–589.
- Kappel F, Granger A, Hrotkó K, Schuster M. 2012. Fruit breeding: handbook of plant breeding. New York (NY): Springer. Chapter 13, Cherry; p. 459–504
- Nguyen L, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Piaskowski J, Hardner C, Cai L, Zhao Y, Iezzoni AF, Peace C. 2018. Genomic heritability estimates in sweet cherry reveal non-additive genetic variance is relevant for industry-prioritized traits. BMC Genet. 19:23.
- Shirasawa K, Isuzugawa K, Ikenaga M, Saito Y, Yamamoto T, Hirakawa H, Isobe S. 2017. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 24:499–508.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45:W6–W11.
- Yuan Z, Fang Y, Zhang T, Fei Z, Han F, Liu C, Liu M, Xiao W, Zhang W, Wu S, et al. 2018. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol J. 16:1363–1374.