
Abstract
In this study, the complete chloroplast (cp) genome of Sophora tonkinensis Gagnep. was determined through Illumina sequencing method. The complete chloroplast genome of S. tonkinensis was 155,640 bp in length and contained a pair of IR regions (25,925 bp) separated by a small single copy region (18,205 bp) and a large single copy region (85,585 bp). The cp genome of S. tonkinensis encoded 127 genes including 84 protein-coding genes, 35 tRNA genes, and 8 ribosomal RNA genes. The overall GC content of S. tonkinensis cp genome is 36.4%. By phylogenetic analysis using the maximum-likelihood (ML) method, S. tonkinensis showed the closest relationship with Sophora flavescens.
Sophora tonkinensis Gagnep. (Leguminosae) grows and is cultivated in southern China. Its medicinal part is root, which is called Sophora Root or Shan Dou Gen as a Chinese herb. The rhizomes and roots of S. tonkinensis have been used in China to treat acute pharyngolaryngeal infections and sore throats (Hunseung et al. Citation2014). In this study, we finished the chloroplast genome of S. tonkinensis using next-generation sequencing, aiming to provide more molecular materials to accurately identify Sophora species.
Plant materials of S. tonkinensis Gagnep. sequenced in this study were acquired from medical plants garden in Guiyang University of Traditional Chinese Medicine (26°57'N, 106°72'E). Both plant materials and total genomic DNA, that was extracted from fresh young leaves using cetyltrimethylammonium bromide (CTAB) method, were stored in the Institute of medicinal plant cultivation and processing, which is affiliated to Pharmaceutical College, Guizhou University of Traditional Chinese Medicine.
For high-throughput sequencing (NGS), paired-end library from DNA extracts was prepared with a NEBNext Library building kits, following manufacturer’ s protocol. Then, the library was sequenced on an Illumina HiSeq2500 platform. After reads quality filtration, the clean reads were assembled by SPAdes 3.11.0 (Bankevich et al. Citation2012). We used the chloroplast genome of S. alopecuroides (accession NO.: MF156140) as a reference sequence to align the contigs and identify gaps. To fill the gap, Price (Ruby et al. Citation2013) and MITObim v1.8 (Hahn et al. Citation2013) were applied and Bandage (Wick et al. Citation2015) was used to identify the borders of the IR, LSC, and SSC regions. The complete sequence was primarily annotated by Plann (Huang et al. Citation2015) combined with manual correction. All tRNAs were confirmed using the tRNAscan-SE search server (Lowe and Eddy Citation1997). Other protein-coding genes were verified by BLAST search on the NCBI website (http://blast.ncbi.nlm.nih.gov/), and manual correction for start and stop codons was conducted. This complete chloroplast genome sequence together with gene annotations were submitted to GenBank under the accession numbers of MH779853.
The chloroplast genome of S. tonkinensis Gagnep. is a typical quadripartite structure with a length of 155,640 bp. The whole cp genome contains a large single-copy (LSC) region of 85,585 bp, a small single-copy (SSC) region of 18,205 bp, and two inverted repeats (IRs) regions of 25,925 bp. The cp genome possesses 127 genes, including 84 protein-coding genes, 8 ribosomal RNA genes (4 rRNA species), and 35 tRNA genes (30 tRNA species). The overall GC content of the cp genome is 36.4%. The genome structure, gene order, and GC content are similar to those of S. flavescens cp genome.
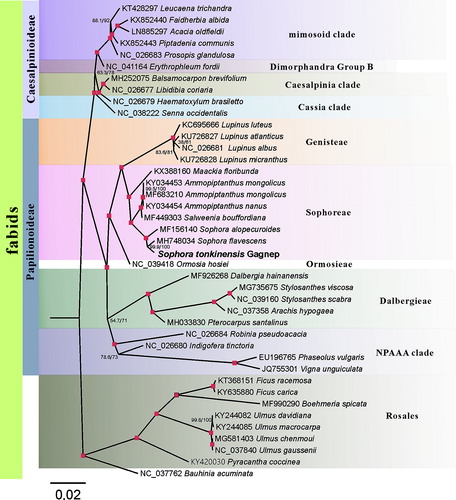
For phylogenetic analysis assessing the relationship of this plastid, we selected other 40 fabids cp genomes to construct a genome-wide alignment. We took plastids of the Rosales clade as the outgroup. The genome-wide alignment of all cp genomes was done by HomBlocks (Guiqi et al. Citation2017), resulting in 47,186 positions in total. The whole genome alignment was analyzed by IQ-TREE version 1.6.6 (Nguyen et al. Citation2015) under the GTR + F+R4 model. The tree topology was verified under both 1000 bootstrap and 1000 replicates of SH-aLRT test. As shown in , the phylogenetic positions of these 41 cp genomes were successfully resolved with full bootstrap supports across almost all nodes. Sophora tonkinensis Gagnep. belongs to the Sophoreae clade as expected, and exhibited the closest relationship with S. flavescens.
Figure 1. Phylogenetic tree yielded by maximum-likelihood (ML) analysis of 41 fabids cp genomes. Maximum-likelihood consensus tree is shown with bootstrap supports indicated by numbers besides branches. Fully resolved nodes were labeled by pink colour.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19:455–477.
- Guiqi B, Mao Y, Xing Q, Cao M. 2017. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110:18–22.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads-a baiting and iterative mapping approach. Nucl Acids Res. 41:e129–e129.
- Huang Daisie I, Quentin CB Cronk. 2015. Plann: a command-line application for annotating plastome sequences. Applications in Plant Sciences. 3:1500026.
- Hunseung Y, Chae H-S, Kim Y-M, Kang M, Ryu KH, Ahn HC, Yoon KD, Chin Y-W, Kim J. 2014. Flavonoids and arylbenzofurans from the rhizomes and roots of Sophora tonkinensis with IL-6 production inhibitory activity. Bioorg Med Chem Lett. 24:5644–5647.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res. 25:955–964.
- Nguyen L-T, Schmidt HA, Haeseler Av, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32:268–274.
- Ruby JG, Bellare P, DeRisi JL. 2013. PRICE: software for the targeted assembly of components of (Meta) genomic sequence data. G3: Genes Genomes Genetics. 3:865–880.
- Wick RR, Schultz MB, Schultz J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31:3350–3352.