
Abstract
Nilaparvata lugens, called as brown planthoppers (BPH), is one of important pests on rice. To identify the origin of Korean N. lugens, we completed mitochondrial genome of N. lugens captured in Guangdong province in China. The circular mitogenome of N. lugens is 17,606 bp including 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNAs, and a single large non-coding region of 2424 bp. The base composition was AT-biased (89.5%). 90 single nucleotide polymorphisms (SNPs) and 10 insertions and deletions are identified by comparing with Korean N. lugens. Phylogenetic trees present that Guangdong BPH may not be an origin of Korean BPH based on distance of two mitochondrial genomes.
Nilaparvata lugens (Stål, 1854), called as brown planthoppers (BPH), is one of the important rice pests in rice-cultivated countries, including China and Korea (Sogawa and Cheng Citation1979). Especially, BPHs have been migrated from China to Korea by westerlies, complete two or three generations, and then affect rice in Korea (Kim et al. Citation1985; Saxena and Barrion Citation1985). Eight BPH mitochondrial genomes mostly originated from China except one from Korea (Park et al. Citation2019) were sequenced. Interestingly, five out of eight mitochondrial genomes do not contain putative control region and around 1 kb region between ND2 and CO1, suspecting that they are partial mitochondrial genome. It indicates that more BPH mitochondrial genomes are required to dissect intra-species variation precisely.
Total DNA of N. lugens captured at Guangxi provinces in China (25°27′26.35′N, 110°16′58.17′E) was obtained (Samples were deposited at InfoBoss Herbarium (IN); Choi NJ, INH-00016) and extracted using the DNA extraction method manually (Zymo Research, USA). Sequencing was conducted by HiSeqX (Macrogen Inc., Korea) and de novo assembly and confirmation were done by Velvet 1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser 1.12 (Zhao et al. Citation2011), BWA 0.7.17 (Li Citation2013), and SAMtools 1.9 (Li et al. Citation2009). Geneious R11 11.1.5 (Biomatters Ltd, Auckland, New Zealand) was used to annotate mitochondrial genome based on N. lugens mitochondrial genome (MK590088; Park et al. Citation2019) together with ARWEN for tRNAs (Laslett and Canbäck Citation2008).
Mitochondrial genome of N. lugens (GenBank accession is MK606370) is 17,607 bp, which is between those of N. lugens captured in Korea (MK590088; Park et al. Citation2019) and Guangdong province, China (Choi et al. Citation2019). It contains 13 protein-coding genes (PGCs), 2 rRNAs, and 22 tRNAs. Its nucleotide composition is AT-biased (A + T ratio is 77.1%). The control region, presumably single largest non-coding AT-rich region, is 2,424 bp (AT ratio is 80.7%).
Fourty-seven single nucleotide polymorphisms (SNPs) and 55 insertions and deletions (INDEL) are identified between those of Guangxi and Guangdong BHP mitochondrial genomes, presenting 10 of 13 PGCs have 12 synonymous SNPs (sSNPs) and ND2 and CYTB contain two non-synonymous SNPs (nsSNPs). While, 41 SNPs and 15 INDELs are found between Guangxi and Korea, showing that 19 sSNPs and 2 nsSNPs from ND2 and ND5. In addition, 90 SNPs and 10 INDELs are identified between Guangdong and Korea among which 17 sSNPs and two nsSNPs in ND5 and CYTB. It indicates that mitochondrial genome of Korean BPH (Park et al. Citation2019) may be originated from Guangxi province, China.
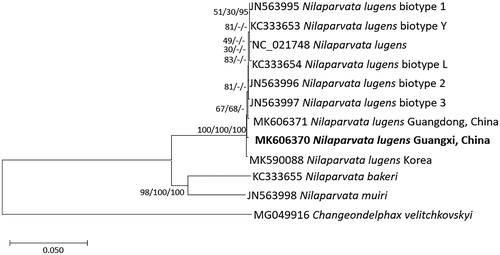
We inferred phylogenetic relationship from 11 Delphacidae complete mitochondrial genomes aligned by MAFFT 7.388 (Katoh and Standley Citation2013). Neighbor joining, minimum evolution, and maximum likelihood bootstrapped trees were constructed using MEGA X (Kumar et al. Citation2018). Phylogenetic trees present that intra-species phylogenetic relationship of N. lugens is not supported by high bootstrap values and some of clades are incongruent among three trees (), indicating that precise analyses of variations from complete mitochondrial genomes together with additional samples from China will be required to understand intraspecific relationship of N. lugens.
Figure 1. Neighbor joining (bootstrap repeat: 10,000), minimum evolution (bootstrap repeat: 10,000), and maximum likelihood (bootstrap repeat: 1,000) phylogenetic trees of 11 Delphacidae and one Fulgoridae complete mitochondrial genomes: Eight Nilaparvata lugens (MK606370 in this study, MK606371, MK590088, JX880069, JN563995, KC333653, JN563996, JN563997, and KC333654), Nilaparvata muiri (JN563998), Nilaparvata bakeri (KC333655), and Changeondelphax velitchkovskyi (MG049916) as an outgroup species. Phylogenetic tree was shown based on neighbor joining tree. The numbers above branches indicate bootstrap support values of neighbor joining, minimum evolution, and maximum likelihood phylogenetic trees, respectively.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Choi NJ, Lee BC, Park J, Park J. 2019. The complete mitochondrial genome of Nilaparvata lugens (Stål, 1854) captured in China (Hemiptera: Delphacidae): investigation of intraspecies variations between countries. Mitochondrial DNA Part B. 4(1):1677–1678.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780.
- Kim Y, Lee J, Park H, Kim M. 1985. Plant damages and yields of the different rice cultivars to brown planthopper (Nilaparvata lugens S.) in fields. Korean J Appl Entomol. 24:79–83.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Laslett D, Canbäck B. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 24:172–175.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv. 13033997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25:2078–2079.
- Park J, Kwon W, Park J, Kim HJ, Lee BC, Kim Y, Choi NJ. 2019. The complete mitochondrial genome of Nilaparvata lugens (stål, 1854) captured in Korea (Hemiptera: Delphacidae). Mitochondrial DNA Part B. 4(1):1674–1676.
- Saxena R, Barrion A. 1985. Biotypes of the brown planthopper Nilaparvata lugens (Stål) and strategies in deployment of host plant resistance. Int J Trop Insect Sci. 6:271–289.
- Sogawa K, Cheng C. 1979. Economic thresholds, nature of damage, and losses caused by the brown planthopper. In: Pathak MD, editor. Brown planthopper: Threat to rice production in Asia. Los Baños, Philippines: International Rice Res Institute; p. 125–142.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829.
- Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 12:S2.