
Abstract
The complete mitochondrial genome of Sarcophyton trocheliophorum was completed using next-generation sequencing (NGS) method. The mitochondrial genome is a circular molecule of 18,508 bp in length, containing 14 protein-coding genes, two ribosomal RNA genes and one transfer RNA gene (Met-tRNA). The base composition is 30.45% A, 16.03% C, 19.13% G, and 34.40% T, with an A + T content of 64.85%. A phylogenetic analysis of Alcyoniidae showed that genus Sarcophyton had the closest relationship with Sinularia.
Species of soft coral genus Sarcophyton are widespread, from Polynesia in the east to the Red Sea in the west. Usually, they habitat in marine environment from the intertidal zone to depths up to 15 m. Colonies of Sarcophyton are characteristically fleshy and soft, mainly mushroom shape with yellow, beige, brown or green colour (Feussner and Waqa Citation2013).
Incomplete mitogenome of Sarcophyton glaucum is the only sequence in genus Sarcophyton, contains 11,715 bp (Beaton et al. Citation1998). In this study, we submitted and analyzed the first complete mitogenome of Sarcophyton, Sarcophyton trocheliophorum (GenBank: MK994517).
An individual of S. trocheliophorum was collected from the South China Sea (West Island, Sanya, Hainan province, China; 18°14′8.75″N, 109°22′39.10″E) and stored in Hainan Tropical Ocean University Museum of Zoology (NO.0001-St). The specimen was identified using mtMutS haplotype similarity. A genomic library was established followed by paired-end (2 × 150 bp) next-generation sequencing (10 Gb), using the Illumina HiSeq X-ten sequencing platform. The quality of produced sequencing reads was checked by FastQC (Andrews Citation2010). The sequences were assembled and mapped to the reference Sinularia mitochondrial genome (Sinularia peculiaris, JX023274) with Spades v3.9.0 (Bankevich et al. Citation2012) and bowtie v2.2.9 (Langmead and Salzberg Citation2012). Protein-coding genes (PCGs) and ribosomal RNA genes (rRNAs) were identified by alignment to the Sinularia peculiaris mitochondrial genome (GenBank: JX023274) and using online server NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html).
The determination of the putative transfer RNA gene (tRNAs) was performed by online software ARWEN (http://130.235.46.10/ARWEN/) and tRNAscan-SE2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/).
The complete mitogenome of S. trocheliophorum was 18,508 in length, with a nucleotide composition of 30.45% A, 16.03% C, 19.13% G and 34.40% T.
The structure of S. trocheliophorum mitogenome was significantly different from classic metazoan mitogenomes, which contain 13 PCGs, 2 rRNAs and 22tRNAs.
The gene content and gene order in present mitogenome are the same as in the other Alcyoniidae, which include 14 PCGs, 2 rRNAs and 1 tRNA. Twelve genes (cox1, 12S, nad1, cytb, nad6, nad3, nad4L, mutS, 16S, nad2, nad5, and nad4) were located on the heavy strand and the other five genes (tRNA-Met, cox3, atp6, atp8 and cox2) were encoded on the light strand. All PCGs were detected to start with the ATG codon. Seven genes (nad1, nad6, nad3, nad2, nad5, cox3 and cox2) appeared to use TAG as stop codon, whereas six genes (cytb, nad4L, mutS, nad4, atp6, and atp8) use the stop codon TAA, and cox1 use no premature stop codon T. We found that there was only one tRNA (Met-tRNA) that can be folded into typical clover-leaf secondary structures. There were a total of 15 gaps between coding genes, the length of which ranged from 3 nucleotides to 112 nucleotides. A 13 nucleotide overlap existed between nad2 and nad5, which was the same as Sinularia peculiaris. The whole mitogenome possessed strong A + T content bias. A + T content of the overall mitogenome was 64.85%, and A + T content of PCGs, rRNAs, and tRNA was 66.26, 58.7, and 56.34%, respectively.
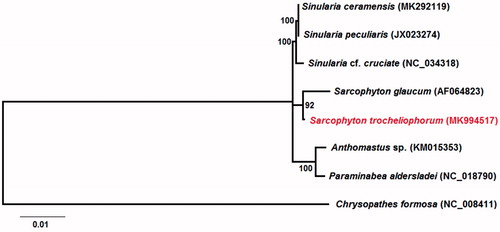
A phylogenetic analysis of family Alcyoniidae was established based on 7 known Alcyoniidae mitogenomes (Beaton et al. Citation1998; Brockman and Mcfadden Citation2012; Kayal et al. Citation2013; Figueroa and Baco Citation2015; Shimpi et al. Citation2017; Asem et al. Citation2019) and an outgroup (Chrysopathes Formosa) (Brugler and France Citation2007). The concatenated dataset for nucleotides contained nine PCGs (published S. glaucum mitogenome lacks nad3, nad4L, nad6, cytb, and mutS) (Beaton et al. Citation1998). The maximum-likelihood (ML) phylogenetic analysis was performed based on the concatenated dataset by using the software MEGA X (Kumar et al. Citation2018). Regarding to phylogenetic tree, genera Sarcophyton and Sinularia revealed close evolutionary relationship ().
Figure 1. Phylogenetic tree of Alcyoniidae based on the concatenated nucleotides of nine protein coding genes and two rRNA genes using maximum-likelihood (ML). Numbers behind each node denote the bootstrap support values.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the manuscript.
Additional information
Funding
Related Research Data
References
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data [accessed 2018 Oct 4]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Asem A, Lu H, Wang P, Li W. 2019. The complete mitochondrial genome of Sinularia ceramensis Verseveldt, 1977 (octocorallia: Alcyonacea) using next-generation sequencing[J]. Mitochondr DNA Part B. 4:815–816.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. Spades: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Biol. 19:455–477.
- Beaton MJ, Roger AJ, Cavalier-Smith T. 1998. Cavalier-smith T. Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: conserved gene order among octocorals. J Mol Evol. 47:697–708.
- Brockman SA, Mcfadden CS. 2012. The mitochondrial genome of Paraminabea aldersladei (cnidaria: Anthozoa: Octocorallia) supports intramolecular recombination as the primary mechanism of gene rearrangement in octocoral mitochondrial genomes. Genome Biol Evol. 4:994–1006.
- Brugler MR, France SC. 2007. The complete mitochondrial genome of the black coral Chrysopathes formosa (cnidaria: Anthozoa: Antipatharia) supports classification of antipatharians within the subclass Hexacorallia[J]. Mol Phylogenet Evol. 42:776–788.
- Feussner K, Waqa T. 2013. Five new species of Sarcophyton (Coelenterata: Octocorallia) from the Fiji Islands. S Pac J Nat App Sci. 31:1–26.
- Figueroa DF, Baco AR. 2015. Octocoral mitochondrial genomes provide insights into the phylogenetic history of gene order rearrangements, order reversals, and Cnidarian phylogenetics. Genome Biol Evol. 7:391–409.
- Kayal E, Roure B, Philippe H, Collins AG, Lavrov DV. 2013. Cnidarian phylogenetic relationships as revealed by mitogenomics. Bmc Evol Biol. 13:5.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with *Bowtie 2. Nat Methods. 9:357.
- Shimpi GG, Vargas S, Poliseno A, Wörheide G. 2017. Mitochondrial RNA processing in absence of tRNA punctuations in octocorals. BMC Mol Biol. 18:16.