
Abstract
The complete mitochondrial genome sequence of Cyathostomum tetracanthum was sequenced in the present study. It was determined to be 13,839 bp bases. The overall base composition was 30.93% A, 45.75% T, 6.97% C, and 16.35% G, with a very strong A + T bias (76.65%). The nucleotide sequence data of 12 protein-coding genes of C. tetracanthum and other 16 Strongylidae species were used for phylogenetic analyses. Cyathostomum tetracanthum was closely related with Cylicocyclus species rather than other Cyathostomum species. The complete mitogenome will facilitate taxonomy and systematics studies of Cyathostominae nematodes.
Strongylidae nematodes, which belongs to Rhabditida, can be further grouped into two subfamilies: Strongylinae and Cyathostominae (Lichtenfels et al. Citation2002). They inhabit the large intestine with a high prevalence in Equus species, which causes a series of clinical symptoms such as anemia, weight loss, and even death. Cyathostomum tetracanthum, which belongs to subfamily Cyathostominae, is a significant Strongylidae nematode. In the present study, the complete mitochondrial genome of C. tetracanthum was generated. Then, phylogenetic analysis of Strongylidae was performed using protein-coding genes in the mitochondrial genome.
Specimen of C. tetracanthum were isolated from the large intestine of infected donkey in Yanjin, Henan Province, China (114°19′E, 33°14′N). The voucher specimen was preserved in 95% ethanol and deposited in the Biological Specimen Museum of Henan Normal University, with an accession number YJ0068. The genomic DNA was extracted from a single specimen using standard phenol/chloroform methods (Sambrook et al. Citation1989). The DNA paired-end library was constructed and sequenced using Illumina Hiseq 4000 platform, then complete mitochondrial genome was assembled with SOAPdenovo v2.04 (Luo et al. Citation2012) and MITObim v1.6 (Christoph et al. Citation2013). The assembled genome was annotated using the DOGMA (Wyman et al. Citation2004) and then submitted into the GenBank database with accession number MN792800.
The complete mitochondrial genome of C. tetracanthum was 13,839 bp in length, which consists of 12 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes, and non-coding regions (NCR), all genes are encoded by the same strand. The overall base composition was 30.93% A, 45.75% T, 6.97% C, and 16.35% G, resulting in a very strong A + T bias (76.65%), in particular, the AT-rich region (84.34%). The size of the 22 tRNAs ranging from 44 to 63 bp. The rrnL (973 bp) is located between trnH and nad3, whereas the rrnS (696 bp) is located between trnE and trnS (UCN).
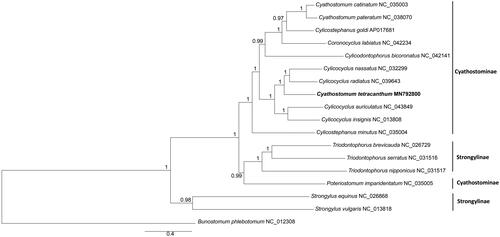
The phylogenetic position of C. tetracanthum was estimated using the Bayesian inference (BI), implemented in MrBayes version 3.1.2 (Ronquist and Huelsenbeck Citation2003). The best-fitting model (GTR + I + G) of sequence evolution for Bayesian analyses was obtained by Modeltest 3.7 (Posada and Crandall Citation1998) under the Akaike Information Criterion. Mitogenome sequences of 16 other Strongylidae species and Bunostomum phlebotomum were retrieved from GenBank. Analyses were performed using 12 protein-coding genes, using B. phlebotomum as outgroup.
Phylogenetic analysis showed that C. tetracanthum was closely related to Cylicocyclus species rather than other Cyathostomum species. Strongylidae nematodes grouped into two major clades: Triodontophorus species and others (). Species of genus Triodontophorus clustered together with Cyathostominae species although they belonged to subfamily strongylinae, which was consistent with previous studies (Gao et al. Citation2017; Li et al. Citation2019). The mitogenome sequences will facilitate taxonomy and systematics studies of Cyathostominae nematodes in the future.
Figure 1. Bayesian 50% majority-rule consensus phylogenetic tree of Cyathostomum tetracanthum and other 16 species Strongylidae nematodes based on 12 protein-coding genes. Bunostomum phlebotomum were used as outgroups.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Christoph H, Lutz B, Bastien C. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 41:e129.
- Gao Y, Zhang Y, Yang X, Qiu JH, Duan H, Xu WW, Chang QC, Wang CR. 2017. Mitochondrial DNA evidence supports the hypothesis that Triodontophorus species belong to Cyathostominae. Front Microbiol. 8:1444.
- Li Q, Gao Y, Wang XX, Li Y, Gao JF, Wang CR. 2019. The complete mitochondrial genome of Cylicocylus ashworthi (Rhabditida: Cyathostominae). Mitochondrial DNA B. 4(1):1225–1226.
- Lichtenfels JR, Gibbons LM, Krecek RC. 2002. Recommended terminology and advances in the systematics of the Cyathostominea (Nematoda: Strongyloidea) of horses. Vet Parasitol. 107(4):337–342.
- Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaSci. 1(1):18.
- Posada D, Crandall KA. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics. 4:817–818.
- Ronquist F, Huelsenbeck JP. 2003. MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19(12):1572–1574.
- Sambrook T, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual. 2nd ed. New York: Cold Spring Harbor Laboratory Press.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.