
Abstract
Myricaria laxiflora (Tamaricaceae) is an endangered species distributed only along the river beach of Yangtze River. Here, the complete chloroplast (cp) genome of M. laxiflora was assembled and characterized. The cp genome is in a total length of 154,971 bp with the typical quadripartite structure of angiosperms, containing 2 inverted repeats (IRs) of 26,004 bp separated by a large single-copy (LSC) region of 84,702 bp and a small single-copy (SSC) region of 18,261 bp. The whole cp genome of M. laxiflora contains 130 genes, including 85 protein-coding genes, 37 tRNAs genes, and 8 rRNAs. Phylogenetic analysis based on the complete cp genomes within the Tamaricaceae family suggests that M. laxiflora is closer to M. paniculata.
Myricaria laxiflora (Franchet) P.Y. Zhang & Y.J. Zhang is an endemic perennial shrub of Tamaricaceae. It is only distributed at the water-level-fluctuation zone within the Three-Gorges Reservoir area of Yangtze River (Chen et al. Citation2005). With the construction of Three Gorges Dam, only few wild populations of this species in Hubei and Sichuan have remained. At present, this species has been Endangered due to habitat loss and the decline in population size (Chen and Wang Citation2015). To conserve this species, many researches mainly focused on reproductive technology and population ecology of this species have been conducted (Chen and Wang Citation2015). In this study, we sequenced and characterized the complete chloroplast (cp) genome of M. laxiflora using Illumina sequencing technology.
Fresh and clean leaves of M. laxiflora were collected from Luzhou, Sichuan, China (N28°45′20.88″, E105°13′33.57″, 224 m). The voucher specimen (qianwang20191204) was deposited in the herbarium of Southwest University (SWCTU). The total genomic DNA was extracted and used for sequencing on Illumina HiSeq 4000 platform at the Beijing Novogene Bioinformatics Technology Co., Ltd. (Nanjing, China). About 2 GB raw data were used to de novo assemble the complete cp genome using SPAdes (Bankevich et al. Citation2012). The complete genome sequence was annotated using PGA (Qu et al. Citation2019) with manual adjustments. The sequence of cp genome was deposited in GenBank (accession numbers MN867948).
The cp genome of M. laxiflora is 154,971 bp in size with the typical quadripartite structure of angiosperms, containing 2 inverted repeats (IRs) of 26,004 bp separated by a large single-copy (LSC) region of 84,702 bp and a small single-copy (SSC) region of 18,261 bp. The total GC content of the whole sequence is 36.3%. The cp genome of M. laxiflora contains 130 genes, including 85 protein-coding genes (PCGs), 37 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes. Seventeen genes including 6 PCGs (ndhB, rpl2, rpl23, rps12, rps7, and ycf2), 7 tRNA genes (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC) and 4 rRNA genes (rrn16, rrn23, rrn4.5, and rrn5) were duplicated. Among the 133 unique genes, 15 had one intron, and 3 had two introns.
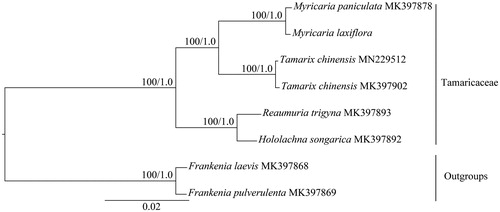
The completed cp genome sequences of M. laxiflora and another eight species belong to Tamaricaceae and Frankeniaceae (outgroups) were used to analyze the phylogenetic position of M. laxiflora. The sequences were aligned with MAFFT (Katoh and Standley Citation2013). The maximum-likelihood (ML) and Bayesian inference (BI) phylogenetic trees were reconstructed using RAxML (Stamatakis Citation2014) and MrBayes (Ronquist et al. Citation2012). The ML and BI analyses generated the same tree topology (). The result showed that the monophyletic Myricaria forms a clade with Tamarix, which is sister to the clade formed by Reaumuria and Hololachna. The complete cp sequence of M. laxiflora reported here will provide a useful resource for the conservation genetics of this species as well as for the phylogenetic studies for Tamaricaceae.
Figure 1. Phylogenetic tree of Tamaricaceae based on eight complete chloroplast genome sequences. Numbers at nodes correspond to ML bootstrap percentages (1000 replicates) and Bayesian inference (BI) posterior probabilities. All the sequences are available in GenBank with the accession numbers listed right to their scientific names.
Disclosure statement
No conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Chen FQ, Wang CH. 2015. Ecological protection of the rare and endangered plant Myricaria laxiflora in the Three Gorges Area. Beijing, China: Science Press (in Chinese).
- Chen FQ, Xie ZQ, Xiong GM, Liu YM, Yang HY. 2005. Reintroduction and population reconstruction of an Endangered plant Myricaria laxiflorain in the Three Gorges Reservoir area, China. Acta Ecol Sin. 25:1811–1817 (in Chinese with English abstract).
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.