
Abstract
Pachira macrocarpa is one popular gardening ornamental plant in East Asia. In this study, we generated the complete chloroplast (cp) genome of P. macrocarpa. The complete cp genome was 157,936 bp in length, containing a large single copy region (LSC) of 88,283 bp, a small single-copy region (SSC) of 21,261 bp, and two inverted repeat (IR) regions of 24,196 bp. The new sequence has a total of 129 genes, including 86 protein-coding genes, 35 tRNA genes, and 8 rRNA genes. Further, phylogenetic analysis showed that P. macrocarpa has a close relationship with Bombax ceiba. This study provides important information for future evolution, genetic and molecular biology studies of P. macrocarpa.
The Pachira macrocarpa Walp belongs to the family Malvaceae. The seed of P. macrocarpa was usually fried to eat, and made into cans (Lin et al. Citation2011). In particular, P. macrocarpa is known as ‘Fa-cai Tree’, which is a popular gardening ornamental plant in East Asia, due to its vibrant, fat, thick leaves that represent wealth and bountiful harvest (Li et al. Citation2009). However, only a few genomic resources have been explored, and the evolutionary relationship of P. macrocarpa is limited in study. Herein, the complete cp genome sequence of P. macrocarpa was deciphered and the phylogenetic relationship with related species was also detected.
Total DNA was extracted from 5 g fresh leaves gathered from Yong’an mountain in Fuyang District, Hangzhou city, Zhejiang province of China (119°55′57″E, 29°52′33″N) using CTAB method (Doyle and Doyle Citation1987) stored in Research Institute of Subtropical Forestry, Hangzhou, China (specimen code: MBLS-009). After DNA quality assessment, sequencing was performed with an Illumina Hiseq 4000 platform (Biodata Biotechnologies Inc., Hefei, China), yielding at least 6.6 GB of clean data for P. macrocarpa. The filtered reads were assembled using the program NOVOPlasty (Dierckxsens et al. Citation2017) with a complete chloroplast genome of its close relative Bombax ceiba as the reference (GenBank accession no. NC037494.1). The chloroplast genes were annotated by DOGMA, using default parameters to predict protein-coding genes, rRNA, and tRNA genes (Wyman et al. Citation2004). Finally, The new annotated complete cp genome of this species had been submitted to NCBI database with an accession number MN894601.
The size of the complete chloroplast genome of P. macrocarpa is 157,936 bp, containing a large single-copy (LSC) region of 88,283 bp, a small single-copy (SSC) region of 21,261 bp, and two inverted repeat (IR) regions of 24,196 bp. The total G + C content of chloroplast genome is 36.91%, and while it is 43.02% of IRs, which is higher than LSC and SSC regions (34.75 and 31.96%, respectively). The new sequence has a total of 129 genes, including 86 protein-coding genes, 35 tRNA genes, and 8 rRNA genes.
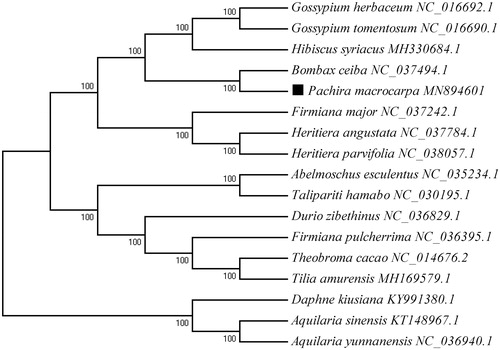
A phylogenetic analysis was carried out using cp genome sequences of P. macrocarpa and other 13 cp genome sequences of species in Malvaceae that could be downloaded from the NCBI database. Meanwhile, three species from the Thymelaeaceae (Daphne kiusiana, Aquilaria sinensis, and Aquilaria yunnanensis) were used as outgroups. The alignment was conducted using MAFFT (Kazutaka and Standley Citation2013). The phylogenetic tree was built by the neighbor-joining (NJ) method using MEGA7 (Kumar et al. Citation2016). The bootstrap analysis with 1000 replicates was used to confirm the stability of each tree node. The results () indicated that P. macrocarpa has a close relationship with Bombax ceiba. The complete cp genome sequence of P. macrocarpa can be great benefit to further study on its population genetics and molecular breeding.
Figure 1. phylogenetic tree inferred from 17 cp genomes. The position of P. macrocarpa is shown with a black box and bootstrapping values are listed for each node.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Kazutaka K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 3(4):772–780.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger dataset. Mol Biol Evol. (7)33:1870–1874.
- Li YH, Ma YM, Han L. 2009. Relationship between the tolerance to dehydration and superoxide dismutases (SODs) in Pachira macrocarpa. Scientia Silvae Sinicae. 45(5):74–79.
- Lin LI, Hong YC, Feng L, Hong HE, Sha Y. 2011. Isolation and identification of the pathogen causing phytophthora blight of Pachira macrocarpa. Acta Horticulturae Sin. 38(12):2395–2400.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. (17)20:3252–3255.