
Abstract
Apple is one of the most important fruit crops in international trade. In this study, we presented the complete mitochondrial genome sequence of apple cultivar ‘Yantai Fuji 8’. The complete mitochondrial genome is 396947 bp in length with an overall GC content of 45.40%. It contains 57 genes including 33 protein-coding genes, 4 rRNAs, and 20 tRNAs. The phylogenetic analysis showed that ‘Yantai Fuji 8’ was clustered with the Malus of Rosaceae family.
The cultivated apple (Malus domestica L.), which belongs to the family Rosaceae, is an iconic tree and a major fruit crop worldwide. Centuries of human exploitation and selection have produced thousands of apple cultivars (Morgan Citation2013). However the parentage of some commerically imported varieties is still unknown because of the incomplete record of breeding (Cornille et al. Citation2014). Molecular comparisons are required to improve our understanding of the diversity and evolutionary history of apple. Mitochondrial data of plants constitute a largely unexplored and potentially rich source of phylogenetic information (Rydin et al. Citation2017). However, more than 90% of all mitochondrial DNA deposited in GenBank is from animals (Smith Citation2016), and data from the mitochondrion have rarely been used to infer phylogeny of plants (Rydin et al. Citation2017). In the present study, the complete mitochondrial genome of apple cv. ‘Yantai Fuji 8’ was determined. We also explored its phylogenetic relationship with other plant species, which would help our better understanding of the evolution of Rosaceae mitochondrial genome.
Specimen of Malus domestica cv. ‘Yantai Fuji 8’ was collected from an experimental orchard in Nanjing Forestry University (N31.603°, E119.1736°) and deposited in Nanjing Forestry University. Total genomic DNA was extracted from the fresh leaves using modified CTAB method. DNA was sheared to construct 500 bp (insert size) paired-end library in accordance with the Illumina HiSeq X platform. Illumina data were filtered by fastp program (Chen et al. Citation2018). Genomic sequence was assembled with NOVOPlasty (Dierckxsens et al. Citation2017), and annotation was performed with the online program GeSeq (Tillich et al. Citation2017). The annotation results were inspected by Geneious 8.0.4 (Kearse et al. Citation2012) and manually adjusted where necessary.The annotated genomic sequence has been submitted to GenBank with the accession number MN964891.
The complete mitochondrial genome has a size of 396947 bp and the overall base composition was 27.3% A, 27.3% T, 22.5% C, and 22.9% G. It consists of 33 protein-coding genes, 4 ribosomal RNA (2x 26S; 18S and 5S) genes and 20 transfer RNA genes including duplication of trnfM(cau) and triplication of the trnF(gaa) genes. All the coding regions accounted for 13.94% of the whole genome. Most of the protein coding genes use the common start codon: ATG, except nad1, which uses GTG as a start codon.
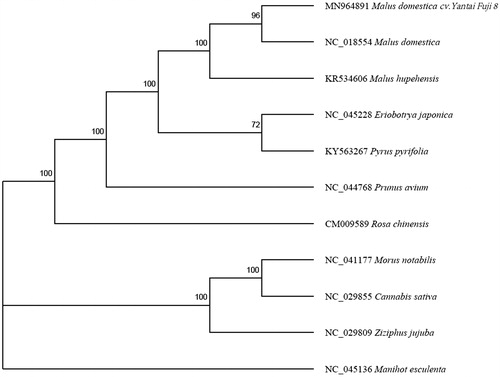
A phylogenetic analysis was performed using the complete mitochondria genome of 10 species within Rosales. Manihot esculenta was used as an outgroup. Multiple sequences alignment was achieved by HomBlocks pipeline (Bi et al. Citation2018). Under the help of the IQ-tree (Lam-Tung et al. Citation2015), Maximum-likelihood analysis was conducted with 1000 replicates of ultrafast bootstrapping. The best-fit model was selected using ModelFinder (Kalyaanamoorthy et al. Citation2017). The phylogenetic tree showed that all Malus species had a close phylogenetic relationship. The availability of the complete mitochondrial genomes will provide additional information for the reconstruction of a phylogenetic model for the Rosaceae species ().
Figure 1. The phylogenetic tree based on the 11 complete mitochondrial genome sequences. Numbers in the nodes are the bootstrap values from 1000 replicates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110(1):18–22.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Cornille A, Giraud T, Smulders MJM, Roldan-Ruiz I, Gladieux P. 2014. The domestication and evolutionary ecology of apples. Trends Genet. 30(2):57–65.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Kalyaanamoorthy S, Bui QM, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. NAT Methods. 14(6):587–589.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lam-Tung N, Schmidt HA, von Haeseler A, Bui QM. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Morgan J. 2013. The new book of apples. New York (NY): Random House.
- Rydin C, Wikstrom N, Bremer B. 2017. Conflicting results from mitochondrial genomic data challenge current views of Rubiaceae phylogeny. Am J Bot. 104(10):1522–1532.
- Smith DR. 2016. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics. 15(1):47–54.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.