
Abstract
Sclerolaena napiformis is an endangered chenopod with a disjunct distribution. Here, we report the complete genomes of two geographically isolated accessions. The two genomes, respectively, were 151,112 and 151,291 bp in length with a pair of inverted repeats (IRs) of 24,345 and 24,339 bp separated by a large single-copy region (LSC) of 83,893 and 84,113 bp and a small single-copy region (SSC) of 18,529 and 18,500 bp, and CG content 36.57% and 36.53%. In total, 130 genes were annotated, including 85 protein-coding genes. Phylogenetic analysis of up to 87 protein-coding genes from 22 plastomes placed the genome of S. napiformis as a sister taxon to the sub-family Salsoloidae within the family Amaranthaceae.
Sclerolaena (Amaranthaceae s.l., subfamily Camphorosmoideae) is a genus of 66 chenopod species endemic to mainland Australia. These perennial herbs or low subshrubs occur mostly in semi-arid areas (VicFlora Citation2016). Hybridization is common among Sclerolaena species and can also occur with species from closely related genera (VicFlora Citation2016). Sclerolaena napiformis Wilson, listed as endangered (Australian Department of the Environment Citation1999), inhabits three disjunct areas in the semi-arid Murray Darling Depression and Riverine regions of southeastern Australia. Precise distribution prior to colonization (<1788) is unconfirmed but S. napiformis has undergone substantial range reduction due to habitat conversion to agriculture (Carta and Parsons Citation2005). Despite fruit characteristics consistent with longer distance dispersal by animals, seed dispersal in other Sclerolaena species is highly localized (Peakall et al. Citation1993) which may contribute to genetic isolation across fragmented populations.
Here, our complete plastid genomes of S. napiformis provide genomic resources to support conservation efforts by enabling assessment of genetic diversity, introgression and phylogeographic patterns.
Fresh leaf material was sampled and vouchered from two cultivated plants grown using seed from the type locality in Jerilderie, New South Wales (MT027237, MEL2470835A, 35°21.68′S, 145°41.21′E) and near Wyuna, Victoria (MT027236, MEL2446779A, 36°12.05′S, 145°06.21′E). Genomic DNA was extracted from silica gel dried material (CTAB protocol for ISOLATE II Plant DNA Kit; Bioline Australia). A genome library was prepared using a TruSeq Nano DNA Library Prep kit (Illumina, San Diego, USA) and sequenced (150 bp PE on an Illumina NovaSeq 6000) at the Australian Genome Research Facility, Parkville, Victoria.
Quality filtering of reads was performed using Trimmomatic 0.39 (Bolger et al. Citation2014); settings: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:150. To recover highly abundant reads from the plastid genome, a highpass filter was performed on filtered reads using BBNorm (sourceforge.net/projects/bbmap/; settings: passes = 1, mindepth = 20,000, target = 999,999,999, minkmers = 110). Recovered reads were error-corrected and normalized using BBNorm using default settings but targeting 50× read depth. Both sets of resulting reads were independently assembled into contigs using the SPAdes genome assembler v3.13.0 (Bankevich et al. Citation2012). Contigs were imported into Geneious R9 (Biomatters Ltd., Auckland, New Zealand) and putative ptDNA contigs were identified using BLASTn comparisons against the Chenopodium quinoa plastid genome (Genbank; MK159176). Assembly was performed via mapping to the C. quinoa reference, manual overlap of contig ends, and by extension of contig ends via multiple rounds of read mapping. Illumina reads were mapped back to the complete plastid contig to confirm accuracy and circularity.
The two complete chloroplast genomes of S. napiformis (MT027236 and MT027237, respectively), were 151,112bp and 151,291bp long with the typical angiosperm structure of two inverted repeats (IRs: 24,345bp, 24,339bp) separated by a large single-copy region (LSC: 83,893bp, 84,113bp) and a small single-copy region (SSC: 18529 bp, 18,500bp) and total CG content of 36.6% and 36.5%. Altogether, 130 genes (plus pseudogene: rpl23; fragment: ycf1 5′) were annotated including 85 protein-coding genes, 37 tRNAs and 8 rRNAs.
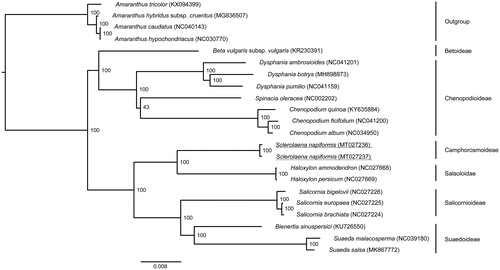
Our phylogeny (), building on Qu et al. (Citation2019), places S. napiformis as sister to Salsoloidae, which is consistent with previous work based on five genetic markers (Kadereit and Freitag Citation2011).
Figure 1. Maximum-likelihood topology depicting the relationship of Sclerolaena napiformis (Genbank: MT027236, MT027237) within the family Amaranthaceae. The analysis was based on an alignment containing 83,421 bp of protein-coding sequence data representing 87 chloroplast genes and three potential pseudogenes from 22 accessions. Members of the genus Amaranthus were used to root the phylogeny. Bootstrap support values are shown at nodes. Analysis was performed using the GTR + G model with 1000 rapid bootstrap iterations implemented in RAxML v8.2.12 (Stamatakis Citation2014). The alignment used to produce this phylogeny is available via Mendeley data (http://dx.doi.org/10.17632/cbvrp8r5z8.1).
Acknowledgements
Thanks to Chris Jenek (RBGV) for maintaining plants in cultivation. Plant material was collected under permits SL102056 (OEH, NSW: MEL2470835A) and 10007717 (DELWP, Vic.: MEL2446779A).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Australian Department of the Environment. 1999. Environment Protection and Biodiversity Conservation Act 1999 (EPBC Act).
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Carta FE, Parsons RF. 2005. Notes on the germination of the endangered species Sclerolaena napiformis (Chenopodiaceae). Cunninghamiana. 9:215–218.
- Kadereit G, Freitag H, 2011. Molecular phylogeny of Camphorosmeae (Camphorosmoideae, Chenopodiaceae): implications for biogeography, evolution of C4-photosynthesis and taxonomy. Taxon. 60(1):51–78.
- Peakall R, Oliver I, Turnbull C L, Beattie AJ. 1993. Genetic diversity in an ant-dispersed chenopod Sclerolaena diacantha. Austral Ecol. 18(2):171–179.
- Qu X-J, Li X-T, Zhang L-Y, Zhang X-J, Fan S-J. 2019. Characterization of the complete chloroplast genome of Suaeda salsa (Amaranthaceae/Chenopodiaceae), an annual succulent halophyte. Mitochondrial DNA Part B. 4(2):2133–2134.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313. doi:10.1093/bioinformatics/btu033. 24451623
- VicFlora. 2016. Flora of Victoria. Royal Botanic Gardens Victoria. [accessed 2019 Dec 6]. https://vicflora.rbg.vic.gov.au.