
Abstract
The mitochondrial genome of a freshwater snail, Cipangopaludina japonica from Nagoya, Central Japan was nearly completely sequenced. This genome encodes 37 genes as found in most other metazoan species. However, there was a changed order of tRNAGlu and tRNAGly genes, a unique feature that was not found in mitochondrial genomes of closely related river snails. Phylogenetic analysis suggested that C. japonica is more closely related to C. ussuriensis from Amur River than to any other Cipangopaludina species sequenced to date.
Cipangopaludina (Bellamya) japonica is a relatively large freshwater snail endemic to Japan. This species can be commonly found in paddy fields, ponds, and swamps throughout the mainland of Japan. It has been a traditional food source in Japan but disappears recently from city areas like Nagoya, Central Japan. The City of Nagoya registers this species as a Vulnerable (VU) species in the RED DATA BOOK NAGOYA 2015 -Animals-. On the other hand, this species was introduced to North America more than 100 years ago and is now known as an invasive alien species, the Japanese Mystery Snail.
On the occasion of the comprehensive survey of freshwater mollusks in Nagoya in 2017, four C. japonica individuals were found from three localities of Nagoya and one of them from the Obata Ryokuchi Park, Moriyama-ku, Nagoya (geographic coordinate: 35.2218 N, 136.9887E) was sequenced in this study. The soft and shell parts of the specimen were both deposited to the Specimen Depository of the Graduate School of Natural Sciences, Nagoya City University with the voucher number SDNCU-A4654.
A small part of mantle muscle was excised, from which DNA was extracted. The mitochondrial genome was amplified as two long PCR fragments and then sequenced using the Illumina MiSeq sequencer. Gap regions were amplified, Sanger-sequenced, and assembled to give rise to a nearly complete mitochondrial genome sequence (16,995 bp, INSD database accession number LC514194) with 37 encoded genes. However, a short region between tRNAGly and tRNAMet genes was never amplified with many combinations of primers and enzymes. The corresponding region amplified and sequenced from the second individual (∼200 bp) included a peculiar sequence comprising of long GT and AT stretches (data not shown), which may have hampered the amplification of this region for the first individual. All protein genes had an ATG start codon except for NADH dehydrogenase subunits 2 and 4 genes which started with ATA. Ten protein genes terminated with a stop codon, whereas the remaining three genes required polyadenylation for the establishment of stop codon in mRNA. We found a gene order change for tRNAGlu and tRNAGly genes in this species, whereas this change was not found for mitogenome sequences of relevant taxa from Chinese localities (Yang et al. Citation2016; Wang et al. Citation2017).
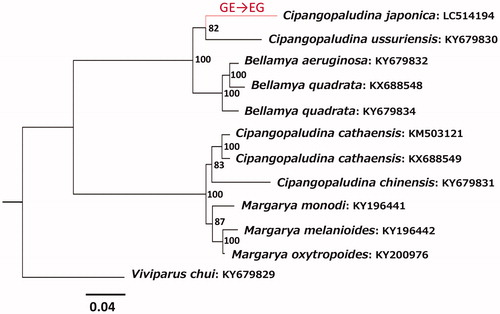
Phylogenetic analysis () suggested that C. japonica is more closely related to C. ussuriensis from Amur River than to any other species with a high bootstrap probability (82%). The clustering of C. japonica and Bellamya (Sinotaia) quadrata with the exclusion of Cipangopaludina (Bellamya) chinensis is consistent with other molecular studies using shorter gene sequences of Japanese individuals (e.g. Hirano et al. Citation2015; Kumazawa et al. Citation2019). Based on the phylogenetic relationships shown in , the gene order change likely occurred after C. japonica diverged from C. ussuriensis. Sequencing of this rearranged region from additional Japanese and Chinese individuals might serve to clarify whether this change in the genome occurred after the ancestor of C. japonica migrated from mainland China.
Figure 1. Phylogenetic position of Cipangopaludina japonica among closely related species and the timing of a gene rearrangement in its mitochondrial genome. A tree shown represents a maximum-likelihood tree constructed using concatenated amino acid sequences of 13 mitochondrial protein genes (3796 sites) with RAxML v8.2.8 (Stamatakis Citation2014) under the mtREV + G substitution model. Numbers at each node are bootstrap probabilities by 1000 replications. INSD accession numbers of mitochondrial genome sequences for each taxon are shown along with the taxon name. The gene order rearrangement for tRNAGlu and tRNAGly genes likely occurred in a terminal lineage leading to C. japonica (shown in red).
Acknowledgments
This study was conducted as a part of the class activity in the Medical School of Nagoya City University for KN supervised by YK. We are grateful to Nagoya Biodiversity Conservation Activity Council and Nagoya Biodiversity Center for collecting and donating freshwater snail samples. We are also grateful to the assistance of the Research Equipment Sharing Center at the Nagoya City University.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Hirano T, Saito T, Chiba S. 2015. Phylogeny of freshwater viviparid snails in Japan. J Mollus Stud. 81(4):435–441.
- Kumazawa Y, Suzuki-Matsubara M, Yokoyama Y, Teramoto T, Murase S, Nasu K, Sun Y, Moriyama A, Kawase M. 2019. DNA barcoding of freshwater molluscs in Nagoya. Bull Nagoya Biodiver Center. 6:1–14 [in Japanese].
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wang J-G, Zhang D, Jakovlić I, Wang W-M. 2017. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). PLoS One. 12(7):e0181699.
- Yang H, Zhang JE, Luo H, Luo M, Guo J, Deng Z, Zhao B. 2016. The complete mitochondrial genome of the mudsnail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(3):1892–1894.