
Abstract
The complete chloroplast genome sequence of a hemiparasitic plant Santalum boninense (Santalaceae) was determined and described in this study. The chloroplast genome was 144,263 bp in length, consisting of a large single-copy region (83,912 bp), a small single-copy region (11,349 bp), and inverted repeats (24,501 bp). The chloroplast genome contained 124 genes including 72 protein-coding genes, 35 tRNA genes, eight rRNA genes, and nine pseudogenes. All the ndh genes and infA gene were found to lose function by gene loss or pseudogenization. GC content of the whole chloroplast genome was 38.0%.
Parasitic plants often reduced photosynthetic ability through gene losses from the chloroplast (cp) genome due to the subsequent relaxation of selective constraints (e.g. McNeal et al. Citation2007; Wicke et al. Citation2013). Hence, it is necessary to compare cp genome data, instead of partial sequences for accurate phylogeny reconstruction including parasitic plants. In this study, we determined and described the complete cp genome sequence of Santalum boninense (Nakai) Tuyama (Santalaceae, Santalales, Eudicots), a hemiparasitic plant that is endemic to the Bonin (Ogasawara) Islands, Japan, based on the Illumina pair-end sequencing data.
The leaf sample was collected from the Chichijima Island of the Bonin Islands (27°5′N, 142°12′E). The voucher specimen was deposited in KYO (the voucher number is Nishimura & Takayama 18031601). Total DNA was extracted with Plant Genomic DNA Extraction Mini Kit (Favorgen Biotech Corp., Taiwan) and sequenced by the Illumina Hiseq platform. Approximately, 19 million 150 bp raw pair-end reads were obtained. We trimmed all reads and used the first 100 bp sequences to improve the quality of the following assembly. NOVOPlasty v3.6 (Dierckxsens et al. Citation2016) was used for assembling cp genome of S. boninense with Osyris alba (Santalaceae) complete cp genome (Petersen et al. Citation2015; KT070882) as a reference. We annotated genes with Geseq (Tillich et al. Citation2017) and checked the results of assembly and genes with Geneious Prime 2020.0.3 (https://www.geneious.com). The complete cp genome sequence was submitted to DDBJ with accession number LC522526.
The cp genome sequence of S. boninense was 144,263 bp in length and had a typical quadripartite structure, consisting of a large single-copy (LSC) region of 83,912 bp, a small single-copy (SSC) region of 11,349 bp, and a pair of inverted repeats (IRs) of 24,501 bp. The cp genome contained 124 genes, including 72 protein-coding genes, 35 tRNA genes, eight rRNA genes, and nine pseudogenes (infA gene, six ndh single genes, and duplicated ndhB genes). The other four ndh genes were not found and thought to have been lost. Fourteen genes were duplicated in IRs. The overall GC content of the cp genome was 38.0% and that of LSC region, SSC region, and IRs was 35.9, 31.6, and 43.1%, respectively.
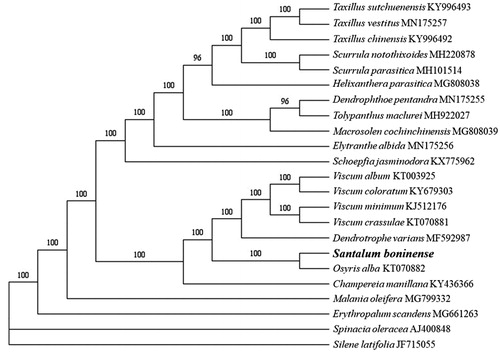
To infer the phylogenetic relationship of S. boninense, we constructed a phylogenetic tree based on the cp genome sequences of S. boninense, other 20 Santalales species, and two Caryophyllales species as the outgroup. All the cp genome data were aligned with MAFFT version 7.450 (Katoh and Standley Citation2013). We constructed maximum-likelihood tree under GTR + GAMMA + I model by RAxML version 8.2.12 (Stamatakis Citation2014). Bootstrap values were calculated with 1000 replicates. The phylogenetic analysis indicated that S. boninense was related to Osyris alba with 100% bootstrap support ().
Figure 1. The maximum likelihood tree based on 21 complete chloroplast genomes of Santalales and two outgroup species. The bootstrap value was shown on each branch.
Acknowledgments
We are grateful to Dr. JiYoung Yang for her advices about the process of cp genome analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2016. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 30(4):772–780.
- McNeal JR, Kuehl JV, Boore JL, de Pamphilis CW. 2007. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 7(1):57.
- Petersen G, Cuenca A, Seberg O. 2015. Plastome evolution in hemiparasitic Mistletoes. Genome Biol Evol. 7(9):2520–2532.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wicke S, Müller KF, de Pamphilis CW, Quandt D, Wickett NJ, Zhang Y, Renner SS, Schneeweiss GM. 2013. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell. 25(10):3711–3725.