
Abstract
Deep-sea fish taxa are elusive, difficult to study, and in multiple cases, represent taxonomic conundrums. Here, we sequenced the complete mitogenome of the shortfin spiny eel, Notacanthus bonaparte, for the first time using low coverage Illumina Paired-end (PE) sequencing. The shortfin spiny eel belongs to the order Notacanthiformes, a relatively unknown taxon of deep-sea marine fishes mostly benthopelagic and bathydemersal. The total length of the assembled mitogenome is 16,623 bp and displays the expected gene orientation for vertebrate mtDNA. Phylogenetic analyses combining all the available mitogenomes of the order Notacanthiformes were performed. The position of N. bonaparte as a member of the Notacanthidae family was confirmed. This mitogenome provides a valuable baseline for future research of N. bonaparte.
The shortfin spiny eel Notacanthus bonaparte Risso, 1840, is a deep-sea marine fish species distributed in the North Atlantic Ocean and the Mediterranean Sea, belonging to the family Notacanthidae (Poulsen et al. Citation2018). There is a general lack of knowledge about the biology of this group of fishes mostly due to the technical challenges and the high costs normally associated with deep-sea exploration. In order to start filling this gap, the mitogenome of Notacanthus bonaparte is described for the first time.
The specimen was captured north of the Iberian Peninsula (44.0847 N; −7.5715 W) at 528.5 meters of depth during the DEMERSALES campaign 2019 and several tissue samples were stored in ethanol. The specimen is stored at the Interdisciplinary Center of Marine and Environmental Research (specimen code NOTAC0008). Genomic DNA was extracted and sent to Macrogen Inc., Korea for whole-genome library construction (350 bp Truseq DNA PCR-free Illumina kit) and sequencing (150 bp Paired-end sequencing on HiseqX150). A 10% subsample of the whole genome sequencing reads was obtained for the mitogenome assembly. Mitogenome assembly and annotation were performed using MITObim v.1.9.1 (Hahn et al. Citation2013) and MitoAnnotator (Wataru et al. 2013), respectively. All available mitogenomes of notacanthiform fishes as well as six outgroups mitogenomes were retrieved from GenBank (accessed in January 2020, ). Individual gene alignments for 13 protein-coding genes (PCG) were produced using GUIDANCE2 server (Sela et al. Citation2015) with MAFFT v7.304 (Katoh and Standley Citation2013) and concatenated using SequenceMatrix v1.7.8 (Vaidya et al. Citation2011), resulting in a final alignment with 11,421 nucleotides. The best partition schemes and molecular evolutionary models were estimated using PartitionFinder2 on XSEDE v2.1.1 (Lanfear et al. Citation2012) and used in phylogenetic analyses.
Figure 1. Bayesian tree of 13 mitogenomes including six notacanthiform species obtained with BI and ML. Node values indicate posterior probabilities and bootstrap supports respectively, with values above 95% represented with asterisks (*). The new mitogenome of N. bonaparte is highlighted in bold.
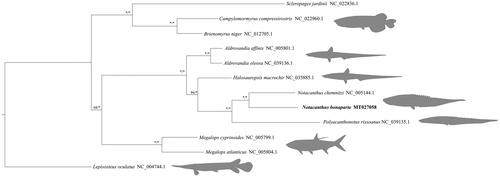
Phylogenetic relationships were estimated using MrBayes on XSEDE v3.2.6 (Ronquist et al. Citation2012) and RAxML-HPC2 Workflow, on XSEDE v8.2.12 (Stamatakis Citation2014). For MrBayes two independent runs were performed (107 generations, sampling one tree for every 1000 generations), each with four chains. For RAxML, 1000 rapid bootstrap replicates and 20ML searches were used. All estimations were performed at CIPRES Science Gateway (https://www.phylo.org) (Miller et al. Citation2010).
The N. bonaparte complete mitogenome has been deposited in GenBank under the accession number MT027058. The total length of the assembled mitogenome is 16,623 bp. Mitogenome length, gene content and orientation are the expected for vertebrate mtDNA: 13 PCGs, 22 transfer RNA (tRNA), 2 ribosomal RNA (rRNA) genes. NAD6 and 8 tRNAs are encoded on the L-strand while the remaining genes are encoded on the heavy strand. The consensus tree obtained from both Maximum-Likelihood (ML) and Bayesian inference (BI) shared an identical topology with higher support values for the BI, (). The mitogenome of N. bonaparte was closely related to the other Notacanthus species present in the tree also clustering inside Notacanthidae family as expected. This information will be useful for further analyses of N. bonaparte.
Acknowledgments
We thank all the participants and crew of the cruise Demersales19 performed on board the R/V Miguel Oliver.
Disclosure statement
The authors declare that there is no conflict of interest.
Additional information
Funding
References
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads - a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Lanfear R, Calcott B, Ho SY, Guindon S. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29(6):1695–1701.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE); Nov 14; New Orleans (LA): GCE. p. 1–8.
- Poulsen J, Thorkildsen S, Arboe N. 2018. Identification keys to halosaurs and notacanthids (Notacanthiformes, Elopomorpha) in the subarctic Atlantic Ocean including three new distributional records and multiple molecular OTUs of Notacanthus cf. chemnitzii. Mar Biodiv. 48(2):1009–1025.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi:10.1093/sysbio/sys029.22357727
- Sela I, Ashkenazy H, Katoh K, Pupko T. 2015. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43(W1):W7–W14. doi:10.1093/nar/gkv318.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Vaidya G, Lohman DJ, Meier R. 2011. Sequence matrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27(2):171–180.
- Wataru I. et al. 2013. MitoFish and MitoAnnotator: A Mitochondrial Genome Database of Fish with an Accurate and Automatic Annotation Pipeline. Mol. Biol. Evol. 30:2531–2540.