
Abstract
The complete chloroplast genome sequence of the Emex australis (GeneBank accession number: MT017652) , the first sequenced of the genus Emex, was reported in this study. The genome size is 159,413 bp in size, consisting of a pair of inverted repeat regions (IR, 30,347 bp), large single copy (LSC, 85,610 bp) and small single copy (SSC, 13,109 bp). This genome contains 113 unique gene including 79 protein coding, 30 tRNA and four rRNA genes. GC content is 37.5%. The maximum likelihood (ML) phylogenetic analysis showed that E. australis was closely related to Rumex acetosa and a sister grouped with genus Rumex.
Genus Emex belongs to Polygonaceae and genus Emex included two species, E. australis Steinh. and E. spinose (L.) Campdera and distributed Africa, southern Europe, and the Middle East (Shivas and Sivasithamparam Citation1994; Schuster et al. Citation2015). E. australis is native to southern Africa and has become a nuturalised species in Australia, California, Hawai, India, Kenya, Newzealand, Pakistan, and Taiwan. (Shivas and Sivasithamparam Citation1994). Turner (1912) reported that E. australis was one of the most obnoxious and aggressive weed in Australia. However, a few researches has been done on E. australis. In this study, we sequenced and analyzed the cp genome of E. australis and it is helpful for future studies on this and another related species.
The sample of E. australis were collected from National Herbarium of Victoria (MEL), Melbourne, Australia (specimen number MEL2393583). Total DNA was extracted using a DNeasy Plant Mini Kit (Qiagen Inc., Valencia, CA, USA) and quantified with a HiGenTM Gel & PCR Purification system (Biofact Inc., Daejeon, Korea). Genomic DNA was sequenced using ana Illumina Hiseq 2500 sequencer (Illumina Inc., San Diego, CA, USA). Paired-end Illumina reads from genomic DNA were assembled de novo with Velvet v. 1.2.08 (Zerbino and Birney Citation2008). The complete chloroplast genome sequence was annotated using Genious R11 (Kearse et al. Citation2012) and Dual Organellar GenoMe annotator (DOGMA) (Wyman et al. Citation2004). tRNAs were identified using the tRNAscan-SE v 1.21 (Schattner et al. Citation2005). A complete chloroplast genome of E. australis was obtained and submitted to GenBank (accession number MT017652).
The cpDNA of E. australis was a duble-stranded circular DNA with 159,413 bp in length, consisting of two inverted repeat (IR) regions of 30,347 bp, a large single-copy (LSC) region of 85,610 bp and a small single-copy (SSC) region of 13,109 bp. The overall GC content of E. australis chloroplast genomes were 37.5%. The GC content of the SSC region (32.2%) was lower than LSC and IR regions (35.6 and 41.2%, respectively). The E. australis cp genome contained 113 genes of which four were ribosomal RNA genes and 30 transfer RNA genes and 79 protein-coding genes. Of 113 genes, 18 genes contained one or two introns.
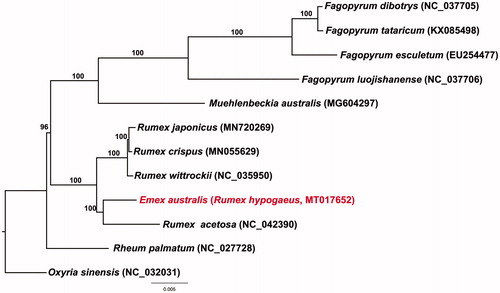
In this study, maximum likelihood phylogenetic analysis was performed by RAxML v 7.4.2 with 1000 bootstraps replicates and the GTR GAMMA model (Stamatakis Citation2006). A total of 77 protein-coding gene sequences of 11 Polygonaceae species (one Emex, one Muehlenbeckia, four Fagonyrum species, five Rumex species) and 1 outgroup (Oxyria sienesis) were aligned using MAFTT (Katoh et al. Citation2002)). This alignment was used for analyses of 64,666 bp. The Polygonaceae was monophyletic and well supported by the bootstrap values (100%). Emex australis was closely related to Rumex acetosa and a sister to other Rumex species ().
Figure 1. A phylogenetic tree of Emex australis and related family in Polygonaceae based on the 77 chloroplast genes of 12 species. The numbers above each branch showed the bootstrap values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server issue):W686–W689.
- Schuster TM, Reveal JL, Bayly MJ, Kron KA. 2015. An updated molecular phylogeny of Polygonaceae (Polygonaceae): relationships of Oxygonum, Pteroxygonum, and Rumex, and a new circumscription of Koenigia. taxon. 64(6):1188–1208.
- Shivas RG, Sivasithamparam K. 1994. Pathogens of Emex australis Steinheil and their potential for biological control. Biocontrol News and Information. 15(3):31N–36N.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bionformatics. 22:2688–2690.
- Wyman S, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo shortread assembly using de Bruijin graphs. Genome Res. 18(5):821–829.