
Abstract
The complete mitochondrial genome of Pealius machili (Hemiptera: Aleyrodidae) is determined to be 15,736 bp in length, comprising 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes, and a putative control region. The overall base composition is 34.60% for A, 10.87% for C, 12.16% for G, and 42.37% for T, with a high AT bias of 76.97%. Eleven reading frame overlaps and 16 intergenic regions were found in the mitogenome of P. machili. The trnS1 and trnS2 lost the dihydrouracil (DHU) arm, while trnL1 lacked the TΨC arm stem. The phylogenetic tree was constructed based on the 13 concatenated PCGs using neighbor-joining method and showed that P. machili is closely related to Aleurochiton aceris, which is in accordance with the traditional classification.
Keywords:
The whitefly Pealius machili belongs to the family Aleyrodidae, which includes around 1556 described species in 161 genera (Martin and Mound Citation2007; Wang et al. Citation2014). Most of these whiteflies are important agricultural pests, and cause serious damage to a wide range of vegetables, flowers, fruits, and green plants (Martin Citation1987). In this study, adult individuals of P. machili were collected from Jianhe County, Guizhou Province, China (N26°38′, E108°29′), and deposited in the insect specimen room of Kaili University with an accession number KLU-Col-2019001.
The complete mitochondrial genome of Pealius machili (GenBank accession number MT015588) is determined to be 15,736 bp in length, comprising 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (rrnL and rrnS), and a putative control region (Boore Citation1999). The overall base composition of P. machili mitogenome is 34.60% for A, 10.87% for C, 12.16% for G, and 42.37% for T, with a high AT bias of 76.97%. The AT-skew and GC-skew of this genome were −0.101 and 0.056, respectively. The gene composition and orientation of P. machili were similar to Aleurochiton aceris mitogenome (GenBank accession number AY572538). Twenty-one genes were transcribed on the minority strand (N-strand), whereas the others were encoded on the majority strand (J-strand).
Gene overlaps in P. machili mitogenome were found in 11 gene junctions and involved a total of 38 bp. The longest overlap was 12 bp in length and located between trnE and trnF. This mitogenome had a total of 159 bp intergenic spacer sequences, which was made up of 16 regions in the range from 1 to 33 bp. The longest intergenic spacer was located between trnS2 and trnR. The control region was located between rrnS and trnI genes with a length of 1337 bp, and the A + T content was 78.01%. The rrnL was located between trnL1 and trnV, and the rrnS between trnV and the control region, respectively. The rrnL was 1199 bp in length with A + T content of 83.65%, and the rrnS was 768 bp in length with A + T content of 83.07%. The length of 22 tRNA genes ranged from 55 bp (trnS2) to 71 bp (trnN and trnM), the A + T content ranged from 67.16% (trnL2) to 91.04% (trnC). Three tRNAs lacked the potential to form the cloverleaf secondary structure. The trnS1 and trnS2 lost the dihydrouracil (DHU) arm, while trnL1 lacked the TΨC arm stem.
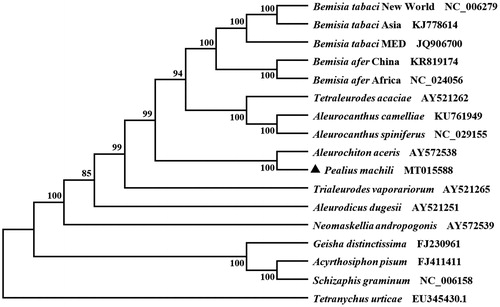
Twelve PCGs started with ATN codons, including four ATAs (nad1, nad2, nad4L, and atp8), four ATTs (cox2, nad4, nad5, and nad6), four ATGs (atp6, cob, nad3, and cox3). However, cox1 used TTG as a start codon. All PCGs were terminated by TAA codon, except for nad5 with a single T instead. According to the relative synonymous codon usage analyses of 13 PCGs, TTT (F), ATT (I), TTA (L), and ATA (M) were the four most frequently used codons. Phenylalanine, isoleucine, leucine, and methionine were the most frequently observed amino acid of 13 PCGs. Based on the concatenated amino acid sequences of 13 PCGs, the neighbor-joining method was used to construct the phylogenetic relationship of P. machili with 15 other whiteflies. The result showed that P. machili is closely related to A. aceris (), which is in accordance with the traditional classification.
Figure 1. Phylogenetic tree showing the relationship between Pealius machili and 15 other whiteflies based on the neighbor-joining method. GenBank accession numbers of each species are listed in the tree. Whitefly determined in this study was labeled with a triangle. Tetranychus urticae was used as an outgroup.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Boore JL. 1999. Survey and summary: animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780.
- Martin JH. 1987. An identification guide to common whitefly pest species of the world (Homopt Aleyrodidae). Trop Pest Managem. 33(4):298–322.
- Martin JH, Mound LA. 2007. An annotated check list of the world’s whiteflies (Insecta: Hemiptera: Aleyrodidae). Zootaxa. 1492(1):1–84.
- Wang JR, Song ZQ, Du YZ. 2014. Six new record species of whiteflies (Hemiptera: Aleyrodidae) infesting Morus alba in China. J Insect Sci. 14:1–5.