
Abstract
In this study, the complete mitochondrial genome of Sarcocheilichthys variegatus wakiyae was sequenced using samples collected from the Tamjin River (type locality), located in Jangheung-gun, Jeollanam-do, Republic of Korea. The complete mitochondrial genome of S. variegatus wakiyae includes 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and a control region (D-loop), with a total length of 16,675 bp. The overall base nucleotide compositions encoded are 30.15% A, 26.71% C, 16.81% G, and 26.33% T with 56.48% A + T. The phylogenetic tree constructed using a neighbor-joining (NJ) method showed that S. variegatus wakiyae and S. variegatus microoculus are not sister groups, despite being subspecies of the same taxonomic species. Rather, S. variegatus wakiyae forms a sister group with S. hainanensis.
Sarcocheilichthys variegatus wakiyae Mori, Citation1927 is an endemic subspecies in the Republic of Korea found in stream flowing to the West and South coasts (Kim and Park Citation2007). This type locality is designated in Choko (Mori Citation1927), which is the former name of the Jangheung-gun. S. variegatus wakiyae was collected from the Tamjin River, which is its type locality, and subjected to phylogenetic analysis. Specifically, the sampling location used in this study was Jangheung-gun, Jeollanam-do, Republic of Korea (34°39'17.0"N 126°53'08.5"E). The samples used for analysis were fixed in 10% formalin solution and stored in the specimen storage facility of Soonchunhyang University (SUC; Voucher No. SUC15576). Genomic DNA was extracted using ventral fin tissue fixed in 99.9% ethanol and polymerase chain reaction experiments were performed. Then, sequencing was performed using the Sanger sequencing method at Macrogen Inc. (Republic of Korea). Next, the sequence data were assembled and annotated using Geneious Prime (Kearse et al. Citation2012).
The complete mitochondrial genome of S. variegatus wakiyae (GenBank Accession No. MT081359) is 16,675 bp long and includes 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and 1 control region (d-loop). In the NADH dehydrogenase subunit, 6 (nd6) gene and 8 tRNA genes are encoded on the light strand (tRNAGln, tRNAAla, tRNAAsn, tRNACys, tRNATyr, tRNASer, tRNAGlu, and tRNAPro). The overall base composition is 30.15% A, 26.71% C, 16.81% G, and 26.33% T with 56.48% A + T, which is similar to the base content and AT bias of other vertebrate mitogenomes (Saccone et al. Citation1999; Lee and Song Citation2016; Gong et al. Citation2020).
The 12S rRNA (959 bp) is located between tRNAPhe and tRNAVal, and the 16S rRNA gene (1688 bp) is located between tRNAVal and tRNALeu. Of the 13 PCGs, the start codons of 12 PCGs is ATG, and that of the co1 gene is GTG. Eight of these PCGs terminate with incomplete stop codons, T (co2, co3, nd6, and cytb) and TA (nd2, atp6, nd3, and nd4), whereas the remaining five genes end with complete stop codons (TAA or TAG). A control region (1006 bp) is located between tRNAPro and tRNAPhe.
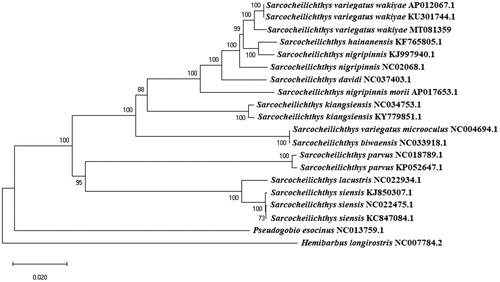
All mitogenome sequences used in the phylogenetic analysis were downloaded from the National Center for Biotechnology Information (NCBI), and subjected to an analysis based on the multiple alignment results obtained by MAFFT ver. 7.450 (Katoh et al. Citation2002; Katoh and Standley Citation2013). The phylogenetic tree was constructed based on the complete mitochondrial genome of species and subspecies in the genus Sarcocheilichthys, and 1000 bootstraps were used when applying the neighbor-joining method, in which the Kimura 2-parameter model (Kimura Citation1980) of MEGA X was used (Kumar et al. Citation2018). Two species belonging to the subfamily Gobioninae were used as outgroups. This implies that S. variegatus wakiyae did not form a sister group with S. variegatus microoculus, which is the same species, but rather is closely related to S. hainanensis in the phylogenetic tree (). The mitogenome collected from this type locality provides an important resource for population genetic analyses and will help researchers to address taxonomic issues.
Figure 1. Phylogenetic tree of the genus Sarcocheilichthys derived from a neighbor-joining (NJ) analysis of the complete mitochondrion genome. The substitution model followed the Kimura 2-parameta (K2P) model. The GenBank accession numbers for each species and subspecies are indicated after the scientific names and only nodes with bootstrap values greater than 70% are shown.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Gong L, Lu Y, Gu L, Dong L, Ren J, Yang S. 2020. Characterization and phylogenetic analysis of the mitochondrial genome of Sarcocheilichthys sinensis (Bleeker) from Baima Hu Lake. Mitochondrial DNA Part B. 5(1):541–542.
- Katoh K, Misawa K, Kuma KI, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Kim IS, Park JY. 2007. Freshwater fishes of Korea. Seoul, Korea: Kyo-Hak Publishing.
- Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 16(2):111–120.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Lee SK, Song HY. 2016. Complete mitochondrial genome of the Korean oily shinner Sarcocheilichthys nigripinnis morii (Cypriniformes, cyprinidae): mitogenome characterization and phylogenetic analysis. Mitochondrial DNA Part B. 1(1):837–838.
- Mori T. 1927. Note on the genus Sarcocheilichthys, with the descriptions of four new species. Annot Zool Japonensis. 11:97–106.
- Saccone C, De Giorgi C, Gissi C, Pesole G, Reyes A. 1999. Evolutionary genomics in metazoa: the mitochondrial DNA as a model system. Gene. 238(1):195–209.