
Abstract
The complete mitochondrial genome of mangrove plant: Aegiceras corniculatum was analyzed in this paper, which is the first for the genus within the family Myrsinaceae. The mitogenome sequence is 425,282 bp in length containing six ribosomal RNA genes, 25 transfer RNA genes, and 39 protein-coding genes. Gene ccmFc, rps3 and rps10 contain one intron, cox2, nad4, nad5 and nad7 contain two introns. Gene nad1 and nad2 are trans-splicing genes. Phylogenetic analysis using the maximum likelihood method revealed that A. corniculatum clustered with Camellia sinensis.
Aegiceras corniculatum belonging to Myrsinaceae species has a wide distribution from Ceylon to South China, through the Malay Archipelago to Polynesia and northeastern Australia (Tomlinson Citation2016). As one important mangrove species, it seems most characteristic of the outer mangal and occurs typically as an isolated low shrub. For the necessarily organs, chloroplast and mitochondria play the important role in higher plant. The whole chloroplast genome of A. corniculatum has been reported (Yan et al. Citation2019) but the Mitogenome is still uncovered. Mitochondrial genome based phylogenetic analysis would improve our understanding of the evolutionary relationship of this plant under tidal habitat. In this study, we sequenced and analyzed the complete mitochondrial DNA sequence of A. corniculatum. This is the first complete mitogenome within the family Myrsinaceae.
Fresh leaves were collected from three individual of A. corniculatum in XinYing Mangrove Natural Garden, Hainan Island (N19°30′, E109°30′), China. The total genomic DNA was extracted from ten mixed fresh leaves of A. corniculatum by using the modified CTAB method (Doyle Citation1987) in the laboratory of Hainan Normal University. Further, the specimen was also stored in the herbarium of Hainan Normal University (No. Sp20190501-001). Genome sequencing was performed on an Illumina Hiseq X Ten platform with paired-end reads of 150 bp. In total, 50.8 Gb short sequence data with Q20 was 96.76% was obtained. The remaining high-quality reads were used to assemble the mitogenome using NOVOPlasty (Dierckxsens et al. Citation2017), where Vaccinium macrocarpon (GenBank accession NC_023338.1) used as the seed sequence. The genes annotation in the mitogenome was doing with SPAdes v.3.9.0 (Bankevich et al. Citation2012) and some genes were annotated manually. The accession number in Genbank is MT130509. Three mitogenome sequences in Primulales were aligned including A. corniculatum, Arabidopsis thaliana and Sesurium portulacastrum were used as the out-group species. Phylogenetic analysis using the maximum likelihood algorithm was conducted with Mega X (Kumar et al. Citation2018).
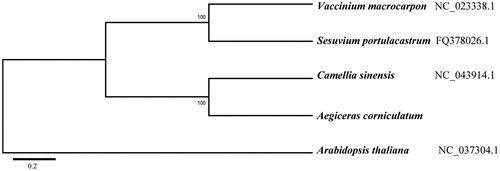
The mitogenome of A. corniculatum is 425,282 bp in length with GC content of 44.82%, which contains three ribosomal RNA genes (rrn5, rrn18 and rrn26), 25 transfer RNA genes, and 39 protein-coding genes. Among those genes, ccmFc, rps3 and rps10 contain one intron, cox2, nad4, nad5 and nad7 contain two introns. Gene nad1 and nad2 are trans-splicing genes. Phylogenetic analysis with other four plant mitogenomes showed that A. corniculatum is sister to Camellia sinensis (). The useful genomic resources for characterization of genetic diversity of A. corniculatum by the mitogenome will help for the study of evolution mechanism.
Figure 1. Maximum likelihood tree based on the sequences of five complete mitogenomes. Numbers in the nodes were bootstrap values from 1000 replicates. Scale in substitutions per site.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol E. 35(6):1547–1549.
- Tomlinson PB. 2016. The Botany of mangrove. Cambridge: Cambridge University Press; p. 302–308.
- Yan XK, Liu TJ, Yuan X, Xu Y, Yan HF, Hao G. 2019. Chloroplast genomes and comparative analyses among thirteen Taxa within Myrsinaceae s.str. Clade (Myrsinoideae, Primulaceae). IJMS. 20(18):4534.