
Abstract
The complete chloroplast genome of Argentina phanerophlebia is reported in this study. The chloroplast genome of A. phanerophlebia was a circular form of 155,565 bp in length. The genome presented a typical quadripartite structure consisting of a pair of inverted repeats (IRa and IRb) of 25,711 bp separated by a large single-copy (LSC) region of 85,691 bp and a small single-copy (SSC) region of 18,452 bp. The genome encoded a set of 129 genes, comprising 84 protein-coding genes, 37 tRNA genes, and eight rRNA genes. Phylogenetic analysis showed that A. phanerophlebia was sister to A. micropetala.
Argentina phanerophlebia (T. T. Yü et C. L. Li) T. Feng et H. C. Wang (synonym: Sibbaldia phanerophlebia T. T. Yü et C. L. Li; Rosaceae Juss.: Potentilleae Sweet) is native to Xizang and Yunnan of China (Li et al. Citation2003). The complete chloroplast (cp) genome of A. phanerophlebia reported herein is useful for further studies on its taxonomy and population genetics.
Fresh leaves of A. phanerophlebia were collected from Huize county, Yunnan Province, China. Voucher specimen (no. Li QQ 20160809017) was deposited in the herbarium of Inner Mongolia Normal University (NMTC). Total genomic DNA was extracted using the CTAB protocol of Doyle and Doyle (Citation1987). Short-insert library (insert size, 300 bp) was prepared and then sequenced using an Illumina HiSeq 2000 Sequencing System in Novogene (Nanjing, China). Illumina paired-end sequencing generated 39,608,674-bp raw reads after adapters were removed. The raw reads were used to assemble the cp genome in NOVOPlasty (Dierckxsens et al. Citation2017) with ribulose-1,5-bisphosphate carboxylase/oxygenase (rbcL) gene from A. micropetala (GenBank accession no. KY420021) as the seed. Chloroplast genome annotation was performed using transferring annotations in Geneious Prime (Kearse et al. Citation2012), with the cp genome of Farinopsis salesoviana (Steph.) Chrtek et Soják (GenBank accession no. MT017928) as the reference. Gene boundaries were manually checked to match the start and stop codons and intron/exon boundaries. The annotated cp genome of A. phanerophlebia was deposited in the GenBank (accession no. MT114192). The cp genome of A. phanerophlebia was a circular form with a size of 155,565 bp in length and had a typical quadripartite structure consisting of a pair of inverted repeats (IRa and IRb: 25,711 bp) separated by a large single-copy (LSC: 85,691 bp) and a small single-copy (SSC: 18,452 bp) regions. The total GC content was 37.1%. The cp genome encoded a set of 129 genes, comprising 84 protein-coding genes, 37 tRNA genes, and eight rRNA genes.
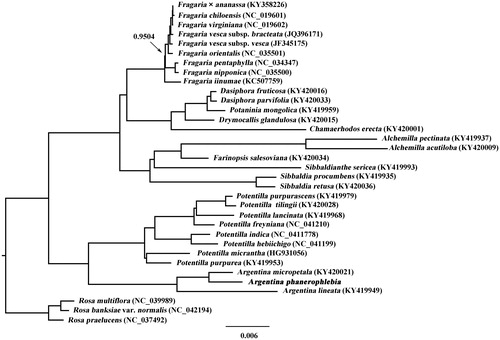
To determine the phylogenetic placement of A. phanerophlebia, the cp genome sequences of 33 Potentilleae and Rosa taxa were downloaded from the GenBank. All the cp genome sequences were aligned with MAFFT version 7.450 (Katoh and Standley Citation2013) and trimmed properly using trimAL version 1.4 (Capella-Gutiérrez et al. Citation2009). The GTR + I + G model was selected as a best-fit model using PartitionFinder2 (Lanfear et al. Citation2016). Bayesian inference (BI) was conducted using the MrBayes version 3.2.2 (Ronquist et al. Citation2012) following Aogan et al. (Citation2020). Phylogenetic tree showed that A. phanerophlebia was sister to A. micropetala ().
Figure 1. Phylogenetic tree resulting from a Bayesian analysis of the cp genome sequences from 31 Potentilleae taxa plus three Rosa taxa as outgroups. Value along branch represents Bayesian posterior probability (only pp < 1.0 is shown).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Aogan, Khasbagan, Li QQ. 2020. The complete chloroplast genome of Farinopsis salesoviana (Rosaceae: Potentilleae). Mitochondrial DNA Part B. 5(2):1363–1364.
- Capella‐Gutiérrez S, Silla‐Martínez JM, Gabaldón T, 2009. trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull. 19(1):11–15.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones‐Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2016. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773.
- Li CL, Ikeda H, Ohba H. 2003. Sibbaldia L. In: Wu ZY, Raven PH, Hong DY, editors. Flora of China. Vol. 9. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press; p. 329–333.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.