
Abstract
Heracleum yungningense is a traditional medicinal plant widely used in China. Here, we reported the complete chloroplast genome of H. yungningense using Illumina paired-end sequencing. The whole genome is 149,223 bp in length, which contains a pair of 18,496 bp inverted repeats regions separated by a large single-copy region and a small single-copy region with 94,770 bp and 17,461 bp in length, respectively. Additionally, the cp genome of H. yungningense contains 113 genes, including 80 protein-coding genes, 29 transfer RNAs, and 4 ribosomal RNAs. The GC content of this cp genome is 37.5%. Phylogenetic analysis was conducted to assess inter-relationships among the major clades of Apiaceae, which strongly supported the close relationship of H. yungningense and H. moellendorffii.
Heracleum yungningense Hand. is an endemic species distributed in western China. It grows under hillside shrubs or on grasses near valleys at an altitude of about 2700 m. Heracleum yungningense is used as H. hemsleyanum, which is a traditional medicinal plant and its root is used to treat numbness in waist and knees, limb cramps, vitiligo, etc. (Wu Citation1988). However, it is threatened by the increasing demands and needs to be protected. Therefore, we here reported the complete chloroplast genome sequence of H. yungningense, and provided the genetic data for the identification and conservation of the species in Heracleum.
Mature leaves of H. yungningense were collected from Xinlong County (30°60′45″N, 100°12′60″E, altitude 3341 m), Sichuan province, China. Voucher specimens (voucher number: ZZY20190920) were deposited in Sichuan University Herbarium (SZ). Genomic DNA was isolated using the modified CTAB method (Doyle Citation1987) and sequenced by Illumina genome analyzer (Hiseq PE150). About 1.5 G clean reads were obtained, which were used to assemble the genome by NOVOplasty v2.7.2 (Dierckxsens et al. Citation2017) with the cp genome of H. moellendorffii (NC 042242; Kang, Yu et al. Citation2019) as the reference. Finally, we used Geneious R9 (v9.0.2) (Kearse et al. Citation2012) to annotate and correct the complete chloroplast genome of H. yungningense according to the other species’ cp genomes published in Apiaceae. A circular genome physical map was drawn using OGDRAW (Lohse et al. Citation2013). The annotated cp genome sequence has been submitted to the GenBank (accession no. MN893285).
The complete cp genome of H. yungningense is 149,233 bp in length, including a large single-copy region of 94,770 bp and a small single-copy region of 17,461 bp separated by a pair of identical inverted repeat regions of 18,496 bp. GC content of the whole genome, IRs, LSC, and SSC regions were 37.5%, 44.6%, 35.9%, and 31.1%, respectively. The cp genome encoded 117 genes, including 80 protein-coding genes, 4 ribosomal RNA genes and 30 transfer RNA genes.
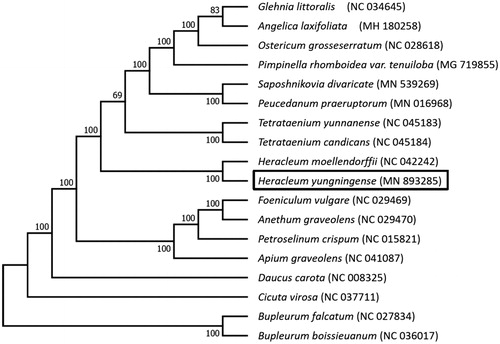
Phylogenetic analysis was conducted to confirm the phylogenetic position of H. yungningense and the inter-relationships among 17 allied species in Apiaceae with Bupleurum falcatum as an outgroup (). All the genome sequences were aligned by MAFFT (Katoh et al. Citation2002) and trimmed in Geneious. Then a maximum likelihood tree was constructed with 1000 bootstrap replicates via RAxML v8.2.4 (Stamatakis Citation2014). As shown in the tree, H. yungningense and H. moellendorffii formed a monophyletic clade with high bootstrap support. T. yunnanense and T. candicans formed a monophyletic clade, which was consistent with previous studies (Kang, Xie et al. Citation2019). Besides, as expected, H. yungningense was closely grouped with T. yunnanense and T. candicans, which were just revised from Heracleum (Xiao et al. Citation2018). The complete cp genome of H. yungningense could subsequently provide informative data for population genomic studies and conservation works of this species.
Figure 1. Maximum-likelihood tree based on the complete chloroplast genomes of 18 species.
Acknowledgments
The authors appreciate people who disclosed cp genomes data in NCBI. The authors thank Xian-Lin Guo, Dan-Mei Su, and Yan-Ping Xiao for their help of sequencing chloroplast genome and data analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucl Acids Res. 45:e18.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Kang L, Xie D, Xiao Q, Peng C, Yu Y, He X. 2019. Sequencing and analyses on chloroplast genomes of Tetrataenium candicans and two allies give new insights on structural variants, DNA barcoding and phylogeny in Apiaceae subfamily Apioideae. PeerJ. 7:e8063.
- Kang L, Yu Y, Zhou SD, He XJ. 2019. Sequence and phylogenetic analysis of complete plastid genome of a medicinal plant Heracleum moellendorffii. Mitochondrial DNA Part B. 4(1):1251–1252.
- Katoh K, Misawa K, Kuma KI, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucl Acids Res. 30(14):3059–3066.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucl Acids Res. 41(W1):W575–W581.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wu ZY. 1988. A compendium of Xinhua materia medica. Shanghai: Shanghai Science and Technology Press.
- Xiao QY, Yu Y, Xie DF, Guo XL, He XJ. 2018. Taxonomic revision of Angelica oncosepala and Heracleum yunnanense. Nord J Bot. 36(3):njb-01563.