
Abstract
Hovenia is a small genus comprising approximately three species and occurs naturally in many countries and regions in Asia. In this study, we assembled and characterized the complete chloroplast genome of H. dulcis and H. trichocarpa for the first time. The two Hovenia chloroplasts have closely resembled gene length, content, and order. The circular genomes of H. dulcis and H. trichocarpa were 161,667 and 161,668 bp in total length, respectively, while each exhibited characteristic quadripartite structure with one LSC region of 89,450–89,451 bp, one SSC region of 18,979 bp, separated by a pair of IR regions of 26,619 bp. A total of 104 unique genes were encoded, including 72 protein-coding genes, 28 tRNA genes, and 4 rRNA genes. Further, the phylogenetic analysis showed that three Hovenia species clustered into one well-supported clade and formed a sister to the genus of Ziziphus.
Hovenia Thunb (Rhamnaceae) is a small genus comprising approximately three species and occurs naturally in many countries and regions in Asia (Chen and Schirarend Citation2000). These species are also cultivated as ornamentals and frequently used for construction, fine furniture, carving, lathework, and musical instruments. Hovenia dulcis is the best-known species, as its swollen pedicels have high sugar content, and a pleasant taste which has been likened to raisins, which may be eaten raw or processed to make beverages. However, relationships within this genus remain poorly resolved. In this study, we reported the complete chloroplast genome sequence of H. dulcis and H. trichocarpa for future phylogenetic studies (GenBank accession number: MT225403 and MT225404).
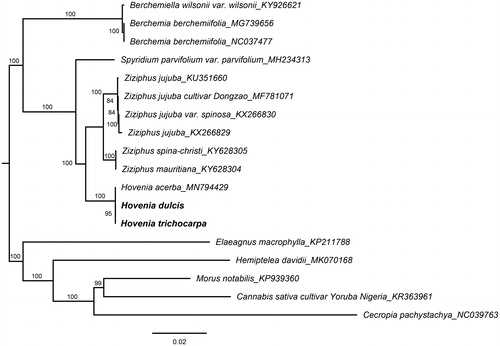
Both voucher specimens were deposited in the Herbarium SYS, including H. dulcis (voucher lxp-10-6092) from Yueyang City, Hunan Province, China (E 113.8640°, N 28.5794°, 200 m a.s.l.) and H. trichocarpa (voucher lxp-13-10048) from the Jiujiang City, Jiangxi Province, China (E 114.9578°, N 29.3817°, 200 m a.s.l.). Total genomic DNA was extracted following the modified CTAB protocol (Yang et al. Citation2014), then sequenced using the Illumina HiSeq2500 platform. The generated reads were assembled by NOVOPlasty (Dierckxsens et al. Citation2017) with H. acerba (MN794429) (Zhang et al. Citation2020) as the reference. The genome annotation was performed with CpGAVAS (Liu et al. Citation2012), then manually adjusted and confirmed the inverted repeat (IR) boundaries using Geneious Prime 2019.2 (https://www.geneious.com/). All available complete chloroplast sequences from the family Rhamnaceae were downloaded from GenBank and aligned with our two Hovenia chloroplasts with MAFFT v7.307 (Katoh and Standley Citation2013). Several species from other families, such as Morus notabilis and Elaeagnus macrophylla, were included as outgroups. The best-fit substitution model was found to be GTR + F+R2 through IQTREE 1.6.12 (Nguyen et al. Citation2015). A maximum-likelihood (ML) phylogenetic tree () was generated with this model using RAxML-HPC BlackBox (Stamatakis Citation2014) by 1000 bootstrap replicates.
Figure 1. Phylogenetic tree reconstruction of 18 taxa with ML method based on complete chloroplast genome sequences. Bootstrap values based on 1000 replicates were provided near branches. GenBank accessions are provided after underlines. H. dulcis and H. trichocarpa are highlighted in bold.
The two new Hovenia chloroplasts have closely resembled gene length, content, and order. The complete chloroplast genome of H. dulcis and H. trichocarpa were 161,667 and 161,668 bp in total length with the overall GC content of 36.7%, respectively. Each genome exhibited characteristic quadripartite structure with one large single- copy (LSC) region of 89,450–89,451 bp, one small single-copy (SSC) region of 18,979 bp, separated by a pair of IR regions of 26,619 bp. A total of 104 unique genes were encoded, including 72 protein-coding genes, 28 tRNA genes, and four rRNA genes, while 18 genes duplicated in the IR regions.
To identify the phylogenetic position of H. dulcis and H. trichocarpa in Rhamnaceae family, an ML tree was constructed based on 11 complete chloroplast genomes from other Rhamnaceae taxa and five outgroups. The results showed that three Hovenia species clustered into one well-supported clade and formed a sister to the genus of Ziziphus (). These newly characterized chloroplast genomes of Hovenia not only could be used to develop highly variable markers for studies on species identification and boundaries, but also for further the phylogeny and evolution of the family Rhamnaceae.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Chen YL, Schirarend C. 2000. Rhamnaceae. In: Wu ZY, Raven PH, editors. Flora of China. Vol. 12. Beijing, China: Missouri Botanical Garden Press; p. 115–119.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de Novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Liu C, Shi LC, Zhu YJ, Chen HM, Zhang JH, Lin XH, Guan XJ. 2012. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13(1):715.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Yang JB, Li DZ, Li HT. 2014. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol Ecol Resour. 14:1024–1031.
- Zhang L, Mao RL, Bi HT, Shen JM, Wang YH, Li MW. 2020. Characterization of the complete chloroplast genome of Hovenia acerba (Rhamnaceae). Mitochondrial DNA B. 5(1):934–935.