
Abstract
Sesbania cannabina (Retz.) Poir. (Fabaceae) is an annual herb, widely distributed in the tropical and subtropical regions. In this study, the complete chloroplast (cp) genome of S. cannabina was reported and phylogenetic analysis was conducted with Fabaceae species based on the cp genome sequences. The cp genome of S. cannabina is 156,894 bp in length, consisting of a pair of inverted repeats (IR, 25,465 bp), a small single-copy (SSC, 18,837bp) and a large single-copy (LSC, 87128 bp) region. It encodes 126 genes including 81 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The Maximum Likelihood tree shows S. cannabina which is nested in Hologalegina clade of the subfamily Papilionoideae (Fabaceae).
The genus Sesbania Adanson (Fabaceae) is usually distributed in tropical and subtropical regions which included around 85 species (Farruggia et al. Citation2018). Most Sesbania species are quite unusual because they prefer the wetland habitats compared to the mostly dry-site inhabiting members of the legume family. Sesbania cannabina (Retz.) Poir (Fabaceae) is an annual herb with strong rhizomes, and it is a source of materials widely cultivated as a fiber plant, especially in India, and it is often used as manure. The sample in this study was collected from Guangzhou, China. The voucher specimen (Duan2019001) was deposited at the South China Botanical Garden (IBSC).
Total genomic DNA were extracted from leaf tissue samples preserved in silica gel using the CTAB method (Doyle Citation1987). Total genomic DNA sample was fragmented into around 500 bp in size. The libraries were prepared and sequenced on the Illumina Hiseq X-Ten plat-form (Illumina Inc., San Diego, CA) with 150 bp paired-end reads. The adapters of the raw data were removed by Trimmomatic (Bolger et al. Citation2014). SPAdes v. 3.11 (Bankevich et al. Citation2012) was applied for the complete chloroplast (cp) genome de novo assembly. The cp genome was annotated by Dual Organellar GenoMe Annotator (DOGMA) (Wyman et al. Citation2004). The cp genome of S. cannabina (GenBank accession no. MT120811) is 156, 894 bp in length, which included a large single-copy (LSC, 87,128 bp), a small single copy (SSC, 18,837 bp), and a pair of inverted repeats (IRs, 25,465 bp). Total 126 genes were annotated, including 81 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The overall G/C content is 35.4%.
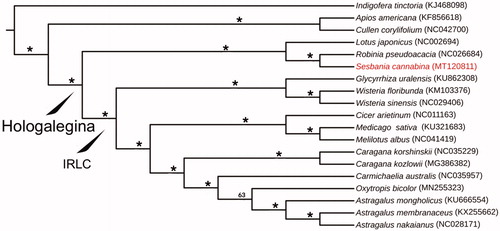
To understand the phylogenetic relationship of this newly sequenced S. cannabina with related genera, totally 18 chloroplast genomes downloaded from GenBank were applied to construct the systematic relationships. The download genomes accession numbers are shown in . We used Geneious prime and Mauve plugin to adjust gene order and remove one copy of IR regions of chloroplast genomes, MAFFT v.7 (Katoh and Standley Citation2013) to align these 19 cp genomes, TrimAl (Capella-Gutiérrez et al., Citation2009) with parameter 0.75 to trim the alignment, and the program IQ-TREE v.1.4.2 (Nguyen et al. Citation2015) with 1000 bootstrap replicates to run a Maximum Likelihood (ML) tree. GenBank accession numbers are given in . The ML tree () shows that the genus Sesbania has a close relationship with Robinia, and they belong to the clade of Hologalegina, which is congruent with the previous study (Farruggia et al., Citation2018).
Figure 1. Maximum-likelihood phylogenetic tree inferred from 19 chloroplast genomes of Fabaceae. The position of Sesbania cannabina is highlighted. The bootstrap values are shown above each node and the values of 100% are shown with asterisks.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Farruggia FT, Lavin M, Wojciechowski MF. 2018. Phylogenetic systematics and biogeography of the pantropical genus Sesbania (Leguminosae). Syst Bot. 43(2):414–429.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment soft-ware version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.