
Abstract
Haemulidae is a widely diverse and widespread family of New World teleost fish. The Peruvian grunt, Anisotremus scapular, inhabits the South American Pacific coast where its overexploitation for human consumption. Fishing has led to the populational decline of the species and raised the need for conservation and aquaculture developments. Here we describe the complete mitogenome of the Peruvian grunt assembled from an RNA-seq dataset. The mitogenome length is 16,785 bp and, as expected for a vertebrate, composed of 13 PCGs, 22 transfer RNA, 2 ribosomal RNA genes. Phylogenetic reconstruction, including all available 25 Lutjaniformes mitogenomes, places A. scapularis within the family Haemulidae, more precisely within the subfamily Haemulinae. Furthermore, paraphyletic relationships were observed within the other groups represented in the phylogeny. Overall, our results highlight the usefulness of using mitochondrial genomes for phylogenetic analysis in Haemulinae as well as the necessity to increase taxonomic sampling within the group.
The family Haemulidae includes 17 genera (∼ 150 species) of mainly marine fish found in the New World (Nelson et al. Citation2016). The Peruvian grunt, Anisotremus scapularis Tschudi, 1846 is a euriphagic benthopelagic fish, distributed from Ecuador to northern Chile, co-habiting with six other Anisotremus species (Chirichigno and Cornejo Citation2001). The high demand by the consumer has led to drastic declines and assessment plans for its recovery, including aquaculture development. The unequivocal identification of the species and the clarification of its phylogenetic relationships are key to the implementation of these strategies. Thus, we obtained the complete A. scapularis mitogenome for the first time.
A specimen of A. scapularis was captured in the south Peruvian shore (−18.0208 S; −70.8695 W). The individual was immediately dissected and tissues from several organs were stored for subsequent total RNA extraction using Illustra RNA spin kit (GE Healthcare, Little Chalfont, UK). TruSeq stranded mRNA libraries were then built and 150 bp paired-end reads sequenced by Macrogen (South Korea). The specimen tissues are stored at the Interdisciplinary Center of Marine and Environmental Research (specimen code SA001). For obtaining the complete mitogenomes from the RNA-se data we followed Moreira et al. (Citation2016), however, mitochondrial contigs were retrieved from the transcriptome assembly using the mode findmitoscaf from MitoZ (Meng et al. Citation2019). Annotation was also performed in MitoZ and manually validated by comparing predicted protein-coding (PCG), ribosomal RNA (rRNA) and transfer RNA (tRNA) genes with other Lutjaniformes using MEGAX (Kumar et al. Citation2018).
For BI and ML phylogenetic analysis, all complete Lutjaniformes mitogenomes (n = 24) and four outgroup taxa, were retrieved from GenBank (accessed in March 2020). The 13 PCG were aligned using GUIDANCE2 (Sela et al. Citation2015) with MAFFT (Katoh and Standley Citation2013) and then concatenated in SequenceMatrix (Vaidya et al. Citation2011). The final concatenated alignment was 11,419 nt long. Best partition schemes and molecular evolutionary models were selected using PartitionFinder2 on XSEDE (Lanfear et al. Citation2016) and used for phylogenetic analyses using MrBayes on XSEDE (Ronquist et al. Citation2012) (GTR + I + G, GTR + I + G and HKY + G) with two independent runs (107 generations, sampling frequency one tree for 1000 generations) and RAxML-HPC BlackBox (Stamatakis Citation2014) (GTR + I + G). All analyses were implemented in CIPRES (Miller et al. Citation2010).
The first complete mitogenome of the subfamily Haemulinae (Haemulidae), from A. scapularis was deposited in GenBank (MT259025). The mitogenome length (16,785 bp), gene content and orientation are those expected for vertebrate mtDNA: 13 PCGs, 22 transfer RNA, 2 ribosomal RNA genes.
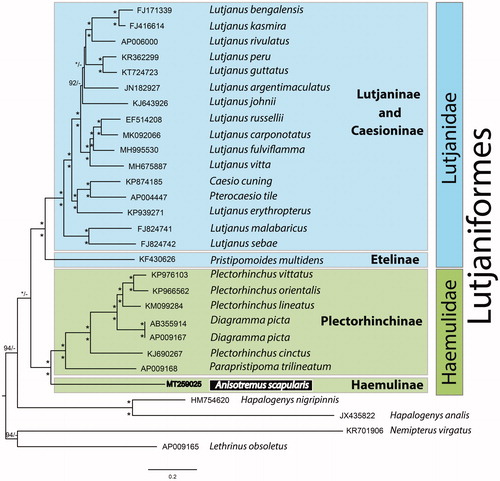
Both BI and ML phylogenetic trees, rooted with the two Spariformes outgroup taxa and for the first time including all available Lutjaniformes mitogenomes in the same analyses, show the same topology (). As expected, Lutjaniformes are divided in two groups corresponding to Lutjanidae and Haemulidae. The latter is further divided in two clades that separate the new mitogenome (i.e. A. scapularis, Haemulinae) from the Plectorhinchinae. The genus Plectorhinchus is paraphyletic regarding Diagramma picta, a result previously observed in a phylogenetic study using both mitochondrial and nuclear markers (Sanciangco et al. Citation2011). Four recent studies using complete mitogenomes for the phylogenetic assessment of the family Lutjanidae (Yamanoue et al. Citation2007; Bayona-Vásquez et al. Citation2017; Zhan et al. Citation2017; Kim et al. Citation2019) revealed conflicting phylogenetic results, probably due to incomplete representation of available mitogenomes. In the present study, by including all available Lutjanidae mitogenomes, we observed that while as expected, Etelinae (i.e. Pristipomoides multidens) clusters neither with Lutjaninae nor with Caesioninae, Lutjaninae appears paraphyletic regarding Caesioninae.
Figure 1. Bayesian Inference phylogenetic tree based on 13 concatenated protein coding genes from 29 mitogenomes, including 25 Lutjaniformes species and four outgroup taxa, two Lobotiformes (HM754620 and JX435822) and two Spariformes (KR701906 and AP009165). Only supports above 92% are shown. The * on the nodes indicate that both posterior probabilities and bootstrap support values above 95%.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that supports the findings of this study are available in NCBI (MT259025) or from the corresponding author, L. Filipe C. Castro.
Additional information
Funding
References
- Bayona-Vásquez NJ, Hernández-Álvarez CA, Glenn T, Domínguez-Domínguez O, Uribe-Alcocer M, Díaz-Jaimes P. 2017. Complete mitogenome sequences of the pacific red snapper (Lutjanus peru) and the spotted rose snapper (Lutjanus gutattus). Mitochondrial DNA Part A DNA. 28(2):223–224.
- Chirichigno N, Cornejo M. 2001. Catálogo comentado de los peces marinos del Perú. Callao (Peru): Instituto del Mar del Perú.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kim G, Lee JH, Alam MJ, Lee SR, Andriyono S. 2019. Complete mitochondrial genome of Spanish flag snapper, Lutjanus carponotatus (Perciformes: Lutjanidae). Mitochondrial DNA Part B. 4(1):568–569.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2016. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol E. 34:772–773.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63–e63.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environments Workshop, GCE 2010. IEEE. p. 1–8. http://ieeexplore.ieee.org/document/5676129/
- Moreira DA, Magalhães MGP, de Andrade PCC, Furtado C, Val AL, Parente TE. 2016. An RNA-based approach to sequence the mitogenome of Hypoptopoma incognitum (Siluriformes: Loricariidae). Mitochondrial DNA Part A. 27(5):3784–3786.
- Nelson JS, Grande TC, Wilson MV. 2016. Fishes of the world. Hoboken (NJ): John Wiley & Sons.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Sanciangco MD, Rocha LA, Carpenter KE. 2011. A molecular phylogeny of the Grunts (Perciformes: Haemulidae) inferred using mitochondrial and nuclear genes. Zootaxa. 2966(1):37–50.
- Sela I, Ashkenazy H, Katoh K, Pupko T. 2015. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43(W1):W7–W14.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Vaidya G, Lohman DJ, Meier R. 2011. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics. 27(2):171–180.
- Yamanoue Y, Miya M, Matsuura K, Yagishita N, Mabuchi K, Sakai H, Katoh M, Nishida M. 2007. Phylogenetic position of tetraodontiform fishes within the higher teleosts: Bayesian inferences based on 44 whole mitochondrial genome sequences. Mol Phylogenet Evol. 45(1):89–101.
- Zhan W, Chen RY, Shen KN, Xu DD, Hsiao C, Der, Lou B. 2017. Next-generation sequencing yields the complete mitochondrial genome of the Redbelly yellowtail fusilier, Caesio cuning (Teleostei: Caesionidae. Mitochondrial DNA Part A DNA Mapping Seq Anal. 28(1):125–126.